GSMR
# Libraries
library(data.table)
library(ggplot2)
library(dplyr)
library(ggrepel)
library(DT)
library(qqman)
library(kableExtra)
library(openxlsx)
library(stringr)
library(gridExtra)
library(reshape2)In this section, we used generalized summary Mendelian randomization
(GSMR)
to test for putative causal associations between vitamin-D and a range
of traits/diseases (e.g. schizophrenia).
Methods
Traits assessed
We calculated the genetic correlation, and performed GSMR analyses, between vitamin D (exposure variable) and the following traits (outcome variables):
- Allergic rhinitis: PCTG in-house analyses of UKB data using BOLT-LMM
- Alzheimer’s disease (AD): Marioni et al. 2018 Transl Psychiatry
- Attention deficit/hyperactivity disorder (ADHD): Demontis et al. 2019 Nat Genet
- Autism spectrum disorder (ASD): Grove et al. 2019 Nat Genet
- Bipolar disorder (BIP): Stahl et al. 2019 Nat Genet
- Coffee intake: PCTG in-house analyses of UKB data using BOLT-LMM
- Coronary artery disease (CAD): van der Harst et al. 2018 Circ Res
- Dyslipidemia: PCTG in-house analyses of UKB data using BOLT-LMM
- Educational attainment (EA): Lee et al. 2018
- Fluid intelligence (IQ): Savage et al. 2018 Nat Genet
- Hypertension: PCTG in-house analyses of UKB data using BOLT-LMM
- Inflammatory bowel disease (IBD): Liu et al. 2015 Nat Genet
- Major depression (MD): Howard et al. 2019 Nat Neurosci
- Parkinson’s disease (PD): Chang et al. 2017 Nat Genet
- Rheumatoid arthritis (RA): Okada et al. 2014 Nat Genet
- Schizophrenia (SCZ): Pardiñas et al. 2018 Nat Genet
- Type II diabetes (T2D): Xue et al. 2018 Nat Commun
Genetic correlations
We ran bi-variate LDSC regression using our vitamin D GWAS results (without correcting for BMI; with BMI as covariate; and conditioned on BMI, i.e. mtCOJO results) and the GWAS summary statistics specified in the ’Traits assessed` tab.
In addition, we uploaded our vitamin D GWAS results to LDHub to get estimates of the genetic correlation with all the traits with GWAS summary statistics available through that web tool.
# ########################################
# LD Hub
# ########################################
# ------ Prepare vitamin D GWAS results for LD Hub
results=$WD/results/fastGWA
ldscDir=$results/ldscFormat
# Main GWAS results (with no adjustment for BMI)
prefix=vitD_fastGWA_withX
cd $ldscDir
echo "snpid A1 A2 b N P-value" | tr ' ' '\t' > $prefix.ldHubFormat
awk '{print $1,$2,$3,$5,$8,$7}' $results/maFormat/$prefix.ma | sed 1d >> $prefix.ldHubFormat
zip $prefix.ldHubFormat.zip $prefix.ldHubFormat
# GWAS results with BMI as covariate
prefix=vitD_BMIcov_fastGWA_withX
cd $ldscDir
zcat $results/$prefix.gz | awk '{print $2,$4,$5,$8,$6,$10}' > $prefix.ldHubFormat
zip $prefix.ldHubFormat.zip $prefix.ldHubFormat
# GWAS results adjusted for BMI (mtCOJO results)
prefix=vitD_BMIcond_fastGWA
cd $ldscDir
echo "SNP A1 A2 b n p" > $prefix.ldHubFormat
sed 1d $results/mtcojo.vitD_fastGWA_condition_on_ukbBMI.mtcojo.cma | awk '{print $1,$2,$3,$9,$8,$11}' >> $prefix.ldHubFormat
zip $prefix.ldHubFormat.zip $prefix.ldHubFormat
# These files then need to be uploaded to the LDHub website
# After downloading the results, edit the output csv file with the rgs so that there is no comma in the name of the following two traits:
## “Offspring birth weight (maternal effect), adjusted for offspring genotype”
## “Own birth weight (fetal effect), adjusted for maternal genotype”
# Otherwise the name is read in two different columns and R is not able to read the results in.
# ########################################
# LDSC
# ########################################
# ------ Format GWAS sumstats for LDSC
# Directories
inDir=$WD/input/gwas_sumstats
results=$WD/results/fastGWA
tmpDir=$results/tmpDir
ldscDir=$bin/ldsc
longdasDir=$medici/longda.jiang/GWAS_sumstats/sumstats_cleaning_Mar2018/cleaned/GSMR_format
# Format GWAS sumstats to munge with LDSC
## Alzheimer's disease
cd $inDir/0_original
echo "SNP A1 A2 b n p" > ../1_LDSRFormat/AD2019
zcat AD_sumstats_Jansenetal.txt.gz | awk -v OFS="\t" '$1=$1' | awk 'NR>1{print $6,$4,$5,$13,$10,$8}' >> ../1_LDSRFormat/AD2019
## Allergic rhinitis
cd $inDir/1_LDSRFormat
sumstats=$inDir/0_original/ukbEUR_ALLERGIC_RHINITIS_cojo.txt
echo "SNP A1 A2 b n p" > ALLERGIC_RHINITIS
awk 'NR>1 {print $1,$2,$3,$5,$8,$7}' $sumstats >> ALLERGIC_RHINITIS
## Coffee intake
cd $inDir/1_LDSRFormat
sumstats=$inDir/0_original/ukbEUR_CI_cojo.txt
echo "SNP A1 A2 b n p" > CI
awk 'NR>1 {print $1,$2,$3,$5,$8,$7}' $sumstats >> CI
## Coronary artery disease
cd $inDir/1_LDSRFormat
sumstats=$longdasDir/CAD_Circulation_Research_2017.txt
echo "SNP A1 A2 b n p" > CAD2017
awk 'NR>1 {print $1,$2,$3,$5,$8,$7}' $sumstats >> CAD2017
## Educational attainment
ln -s $longdasDir/EA_NG_2018_excluding_23andMe.txt $inDir/0_original/EA2018
cd $inDir/1_LDSRFormat
sumstats=$longdasDir/EA_NG_2018_excluding_23andMe.txt
echo "SNP A1 A2 b n p" > EA2018
awk 'NR>1 {print $1,$2,$3,$5,$8,$7}' $sumstats >> EA2018
## Dyslipidemia
cd $inDir/1_LDSRFormat
sumstats=$inDir/0_original/ukbEUR_DYSLIPID_cojo.txt
echo "SNP A1 A2 b n p" > Dyslipidemia2015
awk 'NR>1 {print $1,$2,$3,$5,$8,$7}' $sumstats >> Dyslipidemia2015
## Fluid inteligence
cd $inDir/1_LDSRFormat
sumstats=$medici/uqaxue/GWAS_summary/original/sumstats/IQ_NG_2018.txt
echo "SNP A1 A2 b n p" > IQ2018
awk 'NR>1 {print $1,$2,$3,$5,$8,$7}' $sumstats >> IQ2018
## Hypertension
cd $inDir/1_LDSRFormat
sumstats=$inDir/0_original/ukbEUR_HYPER_cojo.txt
echo "SNP A1 A2 b n p" > hypertension
awk 'NR>1 {print $1,$2,$3,$5,$8,$7}' $sumstats >> hypertension
## Inflammatory bowel disease
cd $inDir/1_LDSRFormat
sumstats=$inDir/0_original/IBD_NG_2015.txt
echo "SNP A1 A2 b n p" > IBD2015
awk 'NR>1 {print $1,$2,$3,$5,$8,$7}' $sumstats >> IBD2015
## MD (PGC)
cd $inDir/0_original
echo "SNP A1 A2 b n p" > ../1_LDSRFormat/7_MDD2019
zcat daner_howard2019.gz | awk 'NR>1 {print $2,$4,$5,log($9),(($17+$18)),$11}' >> ../1_LDSRFormat/7_MDD2019
## Parkinson's disease
cd $inDir/1_LDSRFormat
sumstats=$longdasDir/PD_GWAS_2017.txt
echo "SNP A1 A2 b n p" > PD2017
awk 'NR>1 {print $1,$2,$3,$5,$8,$7}' $sumstats >> PD2017
## Rheumatoid Arthritis
cd $inDir/1_LDSRFormat
sumstats=$longdasDir/RA_NG_2014.txt
echo "SNP A1 A2 b n p" > RA2014
awk 'NR>1 {print $1,$2,$3,$5,$8,$7}' $sumstats >> RA2014
## Type II diabetes
cd $inDir/1_LDSRFormat
sumstats=$medici/uqaxue/data/T2D_meta_summary/Xue_et_al_T2D_META_Nat_Commun_2018_COJO_FORMAT.txt
echo "SNP A1 A2 b n p" > T2D2018
awk 'NR>1 {print $1,$2,$3,$5,$8,$7}' $sumstats >> T2D2018
# Munge GWAS sumstats (process sumstats and restrict to HapMap3 SNPs)
## Outcome traits
cd $inDir/2_LDSRMungeFormat
for i in 7_MDD2019 AD2019 PD2017 RA2014 IBD2015 Dyslipidemia2015 hipertension CI ALLERGIC_RHINITIS T2D2018 IQ2018 CAD2017
do
tmp_command="module load ldsc;
munge_sumstats.py --sumstats $inDir/1_LDSRFormat/$i \
--merge-alleles $ldscDir/w_hm3.snplist \
--out $inDir/2_LDSRMungeFormat/$i"
qsubshcom "$tmp_command" 1 10G munge.$i 10:00:00 ""
done
## Vitamin D
cd $results
for i in vitD_BMIcov_fastGWA vitD_BMIcond_fastGWA
do
tmp_command="module load ldsc;
munge_sumstats.py --sumstats $results/ldscFormat/$i.ldHubFormat \
--merge-alleles $ldscDir/w_hm3.snplist \
--out $results/ldscFormat/$i"
qsubshcom "$tmp_command" 1 30G munge.$i 24:00:00 ""
done
# ------ Run bivariate LDSC with other traits of interest
# Directories
inDir=$WD/input/gwas_sumstats/2_LDSRMungeFormat
results=$WD/results/fastGWA
tmpDir=$results/tmpDir
ldscDir=$bin/ldsc
# Files/settings to use
outcomeList=$WD/input/list_of_traits_LDSC.txt
n_outcomeGWAS=`sed 1d $outcomeList | wc -l`
# Run rg with vitD fastGWA GWAS (vitD, vitD_BMIcov, and vitD_BMIcond)
cd $results/ldsc
for prefix in vitD_fastGWA vitD_BMIcov_fastGWA vitD_BMIcond_fastGWA
do
## Define number of arrays (outcomes) to run
if test $n_outcomeGWAS != 1; then arrays="-array=1-$n_outcomeGWAS"; i="{TASK_ID}"; else arrays=""; i=1; fi
tmp_command="module load ldsc;
outcome_prefix=\$(sed 1d $outcomeList | awk -v i=$i 'NR==i {print \$1}');
outcome_filePath=\$(sed 1d $outcomeList | awk -v i=$i 'NR==i {print \$2}');
ldsc.py --rg $results/ldscFormat/$prefix.sumstats.gz,\$outcome_filePath\
--ref-ld-chr $ldscDir/eur_w_ld_chr/ \
--w-ld-chr $ldscDir/eur_w_ld_chr/ \
--out $results/ldsc/$prefix.\$outcome_prefix.ldsc.rg"
qsubshcom "$tmp_command" 1 30G ${prefix}_biLDSC 24:00:00 "$arrays"
done
GSMR method
Analyses were performed with the GCTA-implemented GSMR method. The following options were used:
- –mbfile: We used a random subset of 20,000 unrelated individuals of European ancestry from the UKB as LD reference (same as previously used for COJO analyses)
- –gsmr-file: The two GWAS used in the analysis:
- Vitamin D GWAS results
- Trait to test causality association with
- –gsmr-direction: We ran bi-directional GSMR analysis (coded as 2)
- –gsmr-snp-min 10 (default): Minimum number of GW
significant and near-independent SNPs required for the GSMR
analysis
- –gwas-thresh 5e-8 (default): P-value threshold to select SNPs for clumping
# #########################################
# Format GWAS results for GSMR
# #########################################
sumstatsDir=$WD/input/gwas_sumstats
longdasDir=$medici/longda.jiang/GWAS_sumstats/sumstats_cleaning_Mar2018/cleaned/GSMR_format
cd $sumstatsDir
## ADHD (PMID 30478444)
## Allele frequencies not provided - use those from UKB EUR
freqs=read.table("UKB_v3EURu_impQC/v3EURu.frq", h=T, stringsAsFactors=F)
df=read.table("gwas_sumstats/0_original/adhd_eur_jun2017.gz", h=T, stringsAsFactors=F)
df$b=log(df$OR)
df$N=53293
df=merge(df, freqs, by="SNP", suffixes=c("","_ukb"))
df=df[df$CHR==df$CHR_ukb,]
df=df[(df$A1==df$A1_ukb & df$A2==df$A2_ukb) | (df$A1==df$A2_ukb & df$A2==df$A1_ukb),]
df$freq=df$MAF
df[df$A1==df$A2_ukb,"freq"]=1-df[df$A1==df$A2_ukb,"MAF"]
df=df[,c("SNP","A1","A2","freq","b","SE","P","N")]
names(df)=c("SNP","A1","A2","freq","b","se","p","n")
write.table(df, "gwas_sumstats/3_maFormat/1_ADHD2017.ma", quote=F, row.names=F)
## Allergic Rhinitis
echo "SNP A1 A2 freq b se p N" > 3_maFormat/ALLERGIC_RHINITIS.ma
awk 'NR>1' 0_original/ukbEUR_ALLERGIC_RHINITIS_cojo.txt >> 3_maFormat/ALLERGIC_RHINITIS.ma
## Alzheimer's disease (PMID 29777097)
echo "SNP A1 A2 freq b se p N" > 3_maFormat/AD2018.ma
awk 'NR>1' $longdasDir/AD_UKB_IGAP_2018May.txt >> 3_maFormat/AD2018.ma
## ASD
# Allele frequencies not provided - use those from UKB EUR
freqs=read.table("UKB_v3EURu_impQC/v3EURu.frq", h=T, stringsAsFactors=F)
df=read.table("gwas_sumstats/fromYeda/3_Original/6_iPSYCH-PGC_ASD_Nov2017", h=T, stringsAsFactors=F)
df=merge(df, freqs, by="SNP", suffixes=c("","_ukb"))
df=df[df$CHR==df$CHR_ukb,]
df=df[(df$A1==df$A1_ukb & df$A2==df$A2_ukb) | (df$A1==df$A2_ukb & df$A2==df$A1_ukb),]
df$freq=df$MAF
df[df$A1==df$A2_ukb,"freq"]=1-df[df$A1==df$A2_ukb,"MAF"]
df$b=log(df$OR)
df$n=46351
df=df[,c("SNP","A1","A2","freq","b","SE","P","n")]
names(df)=c("SNP","A1","A2","freq","b","se","p","n")
write.table(df, "gwas_sumstats/3_maFormat/6_ASD2017.ma", quote=F, row.names=F)
## BIP (PMID 31043756)
setwd("$WD/input")
df=read.table("gwas_sumstats/0_original/daner_PGC_BIP32b_mds7a_0416a.gz", h=T, stringsAsFactors=F)
df$b=log(df$OR)
df$n=df$Nca+df$Nco
df=df[,c("SNP","A1","A2","FRQ_U_31358","b","SE","P","n")]
names(df)=c("SNP","A1","A2","freq","b","se","p","n")
dupSNPs=df[duplicated(df$SNP),"SNP"]
df=df[!df$SNP %in% dupSNPs,]
write.table(df, "gwas_sumstats/3_maFormat/5_BIP2018.ma", quote=F, row.names=F)
## Coffee intake
echo "SNP A1 A2 freq b se p N" > 3_maFormat/CI.ma
awk 'NR>1' 0_original/ukbEUR_CI_cojo.txt >> 3_maFormat/CI.ma
## Coronary artery disease
echo "SNP A1 A2 freq b se p N" > 3_maFormat/CAD2017.ma
awk 'NR>1' $longdasDir/CAD_Circulation_Research_2017.txt >> 3_maFormat/CAD2017.ma
## Dyslipidemia
echo "SNP A1 A2 freq b se p N" > 3_maFormat/Dyslipidemia2015.ma
awk 'NR>1' 0_original/ukbEUR_DYSLIPID_cojo.txt >> 3_maFormat/Dyslipidemia2015.ma
## Educational attainment (PMID 30038396)
echo "SNP A1 A2 freq b se p N" > 3_maFormat/EA2018.ma
awk 'NR>1' $longdasDir/EA_NG_2018_excluding_23andMe.txt >> 3_maFormat/EA2018.ma
## Fluid intelligence (PMID 29942086)
echo "SNP A1 A2 freq b se p N" > 3_maFormat/IQ2018.ma
awk 'NR>1' $medici/uqaxue/GWAS_summary/original/sumstats/IQ_NG_2018.txt >> 3_maFormat/IQ2018.ma
## Hypertension
echo "SNP A1 A2 freq b se p N" > 3_maFormat/hypertension.ma
awk 'NR>1' 0_original/ukbEUR_HYPER_cojo.txt >> 3_maFormat/hypertension.ma
## Inflammatory bowel disease
echo "SNP A1 A2 freq b se p N" > 3_maFormat/IBD2015.ma
awk 'NR>1' 0_original/IBD_NG_2015.txt >> 3_maFormat/IBD2015.ma
## MD (PMID 30718901)
echo "SNP A1 A2 freq b se p N" > 3_maFormat/MDD2019.ma
zcat 0_original/daner_howard2019.gz | awk 'NR>1 {print $2,$4,$5,$7,log($9),$10,$11,(($17+$18))}' >> 3_maFormat/7_MDD2019.ma
## Parkinson’s disease (PMID 28892059)
echo "SNP A1 A2 freq b se p N" > 3_maFormat/PD2017.ma
awk 'NR>1' $longdasDir/PD_GWAS_2017.txt >> 3_maFormat/PD2017.ma
## Rheumatoid Arthritis (PMID 24390342)
echo "SNP A1 A2 freq b se p N" > 3_maFormat/RA2014.ma
awk 'NR>1' $longdasDir/RA_NG_2014.txt >> 3_maFormat/RA2014.ma
## SCZ (PMID 29483656)
setwd("$WD/input/gwas_sumstats")
df=read.table("0_original/clozuk_pgc2.meta.sumstats.txt.gz",h=T,stringsAsFactors=F)
df$A1=toupper(df$A1)
df$A2=toupper(df$A2)
df$SNP=str_split_fixed(df$SNP,":",3)[,1]
df$SNP=as.character(df$SNP)
df=df[!df$SNP %in% c(1:22,"X"),]
df$b=log(df$OR)
df$N=105318
df=df[,c("SNP","A1","A2","Freq.A1","b","SE","P","N")]
names(df)=c("SNP","A1","A2","freq","b","se","p","N")
dupSNPs=df[duplicated(df$SNP),"SNP"]
df=df[!df$SNP %in% dupSNPs,]
write.table(df,"3_maFormat/2_SCZ2018.ma", row.names=F, quote=F)
## Type II Diabetes (PMID 30054458)
echo "SNP A1 A2 freq b se p N" > 3_maFormat/T2D2018.ma
awk 'NR>1' $medici/uqaxue/data/T2D_meta_summary/Xue_et_al_T2D_META_Nat_Commun_2018_COJO_FORMAT.txt >> 3_maFormat/T2D2018.ma
# #########################################
# Generate LD reference dataset
# #########################################
## Extract a random set of 20K unrelated Europeans to use as LD reference
## In addition, keep only the variants in the vittamin D GWAS to speed up GSMR analysis
prefix=vitD_fastGWA
# Directories
results=$WD/results/fastGWA
random20k_dir=$WD/input/UKB_v3EURu_impQC/random20K
## Extract SNPs in fastGWA results
zcat $results/$prefix.gz | awk 'NR>1{print $2}' > $random20k_dir/$prefix.snpList
## Extract data
ld_ref=$medici/UKBiobank/v3EURu_impQC/ukbEURu_imp_chr{TASK_ID}_v3_impQC
ids2keep=$medici/UKBiobank/v3Samples/ukbEURu_v3_all_20K.fam
snps2keep=$WD/input/UKB_v3EURu_impQC/random20K/$prefix.snpList
output=$random20k_dir/ukbEURu_imp_chr{TASK_ID}_v3_impQC_random20k_maf0.01
cd $random20k_dir
plink=$bin/plink/plink-1.9/plink
tmp_command="$plink --bfile $ld_ref \
--keep $ids2keep \
--extract $snps2keep \
--make-bed \
--out $output"
qsubshcom "$tmp_command" 1 10G ukbEURu_v3_impQC_random20k_maf0.01 24:00:00 "-array=1-22"
# #########################################
# Generate GSMR input files
# #########################################
# GSMR takes as input files that have the path names to the datasets to use in the analysis.
# Directories
results=$WD/results/fastGWA
gsmr_inputDir=$results/gsmr/input
random20k_dir=$WD/input/UKB_v3EURu_impQC/random20K
## LD reference data
echo $random20k_dir/ukbEURu_imp_chr1_v3_impQC_random20k_maf0.01 > $gsmr_inputDir/gsmr_ldRef.txt
for i in `seq 2 22`; do echo $random20k_dir/ukbEURu_imp_chr${i}_v3_impQC_random20k_maf0.01 >> $gsmr_inputDir/gsmr_ldRef.txt; done
## Exposure (Vitamin D GWAS)
## Note: Only the 1st file has X chromosome results
echo "vitD $results/maFormat/vitD_fastGWA_withX.ma" > $gsmr_inputDir/gsmr_exposure_vitD
echo "vitD_BMIcov $results/maFormat/vitD_BMIcov_fastGWA.ma" > $gsmr_inputDir/gsmr_exposure_vitD_BMIcov
echo "vitD_BMIcond $results/maFormat/vitD_BMIcond_fastGWA.ma" > $gsmr_inputDir/gsmr_exposure_vitD_BMIcond
## Outcome (Other GWAS)
cat $WD/input/list_of_traits_GSMR.txt | sed 's/ /_/g' > $gsmr_inputDir/gsmr_outcomes.txt
# Split into individual files (for each outcome GWAS to run separately)
cd $gsmr_inputDir
rm gsmr_outcome_*
split -l 1 --numeric-suffixes $gsmr_inputDir/gsmr_outcomes.txt
rm x00
for i in `ls x0*`; do mv $i `echo $i | sed 's/x0/x/'`; done
n=`ls x* | wc -l`
for i in `seq 1 $n`
do
trait=`sed 1d $gsmr_inputDir/gsmr_outcomes.txt | awk -v i=$i 'NR==i {print $1}'`
mv x$i gsmr_outcome_$trait
done
# ########################
# Run bi-directional GSMR
# ########################
results=$WD/results/fastGWA
inDir=$results/gsmr/input
tmpDir=$results/tmpDir
# Files/settings to use
ld_ref=$inDir/gsmr_ldRef.txt
n_outcomeGWAS=`sed 1d $inDir/gsmr_outcomes.txt | wc -l`
# Run GSMR without HEIDI
cd $results
gcta=$bin/gcta/gcta_1.92.3beta3
for exposure in `ls $inDir/gsmr_exposure_*`
do
## Define number of arrays (outcomes) to run
if test $n_outcomeGWAS != 1; then arrays="-array=1-$n_outcomeGWAS"; i="{TASK_ID}"; else arrays=""; i=1; fi
## Define exposure
prefix=`basename $exposure | sed 's/gsmr_exposure_//'`
## Run on all outcomes
tmp_command="outcome=\$(sed 1d $inDir/gsmr_outcomes.txt | awk -v i=$i 'NR==i {print \$1}');
outcomeFile=$inDir/gsmr_outcome_\$outcome;
output=$tmpDir/gsmr_with20kUKB_noHeidi_${prefix}.\$outcome;
$gcta --mbfile $ld_ref \
--gsmr-file $exposure \$outcomeFile \
--gsmr-direction 2 \
--heidi-thresh 0 \
--effect-plot \
--gsmr-snp-min 10 \
--gwas-thresh 5e-8 \
--threads 16 \
--out \$output"
qsubshcom "$tmp_command" 16 30G ${prefix}_gsmr_with20kUKB_noHeidi 24:00:00 "$arrays"
echo ${prefix}_gsmr_with20kUKB_noHeidi
done
cat $tmpDir/gsmr_with20kUKB_noHeidi_*gsmr | head -1 > $results/gsmr/gsmr_with20kUKB_noHeidi.gsmr
cat $tmpDir/gsmr_with20kUKB_noHeidi_*gsmr | grep -v "Exposure" >> $results/gsmr/gsmr_with20kUKB_noHeidi.gsmr
# Run GSMR with new HEIDI-outlier method
cd $results
gcta=$bin/gcta/gcta_1.92.3beta3
for exposure in `ls $inDir/gsmr_exposure_*`
do
## Define number of arrays (outcomes) to run
if test $n_outcomeGWAS != 1; then arrays="-array=1-$n_outcomeGWAS"; i="{TASK_ID}"; else i=1; fi
## Define exposure
prefix=`basename $exposure | sed 's/gsmr_exposure_//'`
## Run on all outcomes
tmp_command="outcome=\$(sed 1d $inDir/gsmr_outcomes.txt | awk -v i=$i 'NR==i {print \$1}');
outcomeFile=$inDir/gsmr_outcome_\$outcome;
output=$tmpDir/gsmr_with20kUKB_withHeidi_${prefix}.\$outcome;
$gcta --mbfile $ld_ref \
--gsmr-file $exposure \$outcomeFile \
--gsmr-direction 2 \
--effect-plot \
--gsmr-snp-min 10 \
--gwas-thresh 5e-8 \
--gsmr2-beta \
--heidi-thresh 0.01 0.01 \
--threads 16 \
--out \$output"
qsubshcom "$tmp_command" 16 30G ${prefix}_gsmr_with20kUKB_withHeidi 24:00:00 "$arrays"
echo ${prefix}_gsmr_with20kUKB_withHeidi
done
cat $tmpDir/gsmr_with20kUKB_withHeidi_*gsmr | head -1 > $results/gsmr/gsmr_with20kUKB_withHeidi.gsmr
cat $tmpDir/gsmr_with20kUKB_withHeidi_*gsmr | grep -v "Exposure" >> $results/gsmr/gsmr_with20kUKB_withHeidi.gsmr
2SMR
To validate causal associations identified with GSMR, we ran 2-sample Mendelian randomization analyses (2SMR). For comparability, we used the same instruments that were used in the GSMR analyses. Note, however, that in some cases the number of instruments is lower in 2SMR because palindromic variants with intermediate allele frequencies are excluded with this method.
# #########################################
# Generate table with all combinations of exposure and outcome GWAS to run 2SMR with
# #########################################
WD=getwd()
# Details of vitamin D GWAS
exposureList=data.frame(Trait=c("vitD","vitD_BMIcov","vitD_BMIcond"),
FilePath=c(paste0(WD,"/results/fastGWA/maFormat/vitD_fastGWA_withX.ma"),
paste0(WD,"/results/fastGWA/maFormat/vitD_BMIcov_fastGWA.ma"),
paste0(WD,"/results/fastGWA/maFormat/vitD_BMIcond_fastGWA.ma")),
stringsAsFactors=F)
# Details of other trait GWAS
outcomeList=read.table("input/list_of_traits_2SMR.txt", h=T, sep="\t", stringsAsFactors=F)
outcomeList$Trait=gsub(" ", "_", outcomeList$Trait)
# Combine in one table
df=cbind(exposureList, outcomeList[rep(1:nrow(outcomeList), each=3),])
tmp=cbind(outcomeList[rep(1:nrow(outcomeList), each=3),], exposureList)
df=rbind(df,tmp)
names(df)=c("exposure","exposureFile","outcome","outcomeFile")
df$heidi="noHeidi"
tmp=df
tmp$heidi="withHeidi"
df=rbind(df,tmp)
df[grep("vitD", df$exposure),"direction"]="12"
df[grep("vitD", df$outcome),"direction"]="21"
# Save table
write.table(df, "results/fastGWA/twosmr/input/analyses2run", quote=F, row.names=F)# #########################################
# Extract instruments from GSMR analyses
# #########################################
results=$WD/results/fastGWA
tmpDir=$results/tmpDir
traitList=$WD/input/list_of_traits_2SMR.txt
# Loop through outcome traits and get instruments used in GSMR analyses
n=`wc -l < $traitList`
for i in `seq 2 $n`
do
# Define outcome trait in this iteration
trait_name=`awk -F"\t" -v i=$i 'NR==i {print $1}' $traitList | tr ' ' '_'`
# Loop through exposure traits and get instruments used in GSMR analyses
for exposure in vitD vitD_BMIcov vitD_BMIcond
do
echo $i $trait_name $exposure
# Get lists of instruments used in both directions - no HEIDI
heidi=noHeidi
gsmr_log=$tmpDir/gsmr_with20kUKB_${heidi}_$exposure.$trait_name.eff_plot.gz
zcat $gsmr_log | tail -4 | sed 1q | tr ' ' '\n' > $tmpDir/gsmr_with20kUKB_${heidi}_$exposure.$trait_name.gsmr12instruments
zcat $gsmr_log | tail -2 | sed 1q | tr ' ' '\n' > $tmpDir/gsmr_with20kUKB_${heidi}_$exposure.$trait_name.gsmr21instruments
# Get lists of instruments used in both directions - with HEIDI
heidi=withHeidi
gsmr_log=$tmpDir/gsmr_with20kUKB_${heidi}_$exposure.$trait_name.eff_plot.gz
zcat $gsmr_log | tail -4 | sed 1q | tr ' ' '\n' > $tmpDir/gsmr_with20kUKB_${heidi}_$exposure.$trait_name.gsmr12instruments
zcat $gsmr_log | tail -2 | sed 1q | tr ' ' '\n' > $tmpDir/gsmr_with20kUKB_${heidi}_$exposure.$trait_name.gsmr21instruments
done
done
# #########################################
# R script to run 2SMR
# #########################################
echo 'args=commandArgs(trailingOnly=T)
WD=args[1]
exposure=args[2]
outcome=args[3]
exposure_GWASfile=args[4]
outcome_GWASfile=args[5]
direction=args[6]
heidi=args[7]
setwd(WD)
library(TwoSampleMR)
# Load exposure sumstats
gwas=read.table(exposure_GWASfile, h=T, stringsAsFactors=F)
## Restrict exposure sumstats to instruments used in GSMR
instruments_filePrefix=ifelse(direction=="12",
paste0("gsmr_with20kUKB_",heidi,"_",exposure,".",outcome,".gsmr",direction,"instruments"),
paste0("gsmr_with20kUKB_",heidi,"_",outcome,".",exposure,".gsmr",direction,"instruments"))
instruments=read.table(paste0("results/fastGWA/tmpDir/",instruments_filePrefix), h=F, stringsAsFactors=F)[,1]
df=gwas[gwas$SNP %in% instruments,]
names(df)=c("SNP","effect_allele","other_allele","eaf","beta","se","pval","samplesize")
df$Phenotype=paste0(exposure,".",heidi)
## Define as exposure
exposure_dat=format_data(df, type="exposure")
# Load outcome sumstats
outcome_dat=read_outcome_data(filename=outcome_GWASfile,
snps=exposure_dat$SNP,
snp_col="SNP",
beta_col="b",
se_col = "se",
effect_allele_col = "A1",
other_allele_col = "A2",
eaf_col = "freq",
pval_col = "p",
samplesize_col = "N")
outcome_dat$outcome=outcome
# Harmonise the exposure and outcome data
dat=harmonise_data(exposure_dat, outcome_dat)
# Perform MR
res=mr(dat)
# Save results
write.table(res, paste0("results/fastGWA/twosmr/",exposure,".",heidi,".",outcome,".2smr"), quote=F, row.names=F)
' > $WD/scripts/run2smr.R
# #########################################
# Submit 2SMR jobs
# #########################################
results=$WD/results/fastGWA
analyses2run=$results/twosmr/input/analyses2run
n=`wc -l < $analyses2run`
cd $results
if test $n != 1; then arrays="-array=2-$n"; i="{TASK_ID}"; else arrays=""; i=1; fi
get_inputParameters="exposure=\$(awk -v i=$i 'NR==i {print \$1}' $analyses2run);
outcome=\$(awk -v i=$i 'NR==i {print \$3}' $analyses2run);
exposure_GWASfile=\$(awk -v i=$i 'NR==i {print \$2}' $analyses2run);
outcome_GWASfile=\$(awk -v i=$i 'NR==i {print \$4}' $analyses2run);
direction=\$(awk -v i=$i 'NR==i {print \$6}' $analyses2run);
heidi=\$(awk -v i=$i 'NR==i {print \$5}' $analyses2run)"
run2smr="Rscript $WD/scripts/run2smr.R $WD
\$exposure
\$outcome \
\$exposure_GWASfile
\$outcome_GWASfile \
\$direction
\$heidi"
qsubshcom "$get_inputParameters; $run2smr" 1 30G run2smr 5:00:00 "$arrays"
# Merge results
sed 1q $results/twosmr/*.2smr | uniq | tr ' ' '\t' > $results/twosmr/all.2smr
cat $results/twosmr/*.2smr | grep -v 'method' | tr ' ' '\t' >> $results/twosmr/all.2smr
# Change method names (no spaces)
sed -i 's/MR\tEgger/MR Egger/g' $results/twosmr/all.2smr
sed -i 's/Weighted\tmedian/Weighted median/g' $results/twosmr/all.2smr
sed -i 's/Inverse\tvariance\tweighted/Inverse variance weighted/g' $results/twosmr/all.2smr
sed -i 's/Simple\tmode/Simple mode/g' $results/twosmr/all.2smr
sed -i 's/Weighted\tmode/Weighted mode/g' $results/twosmr/all.2smr
Results
Genetic correlations
# ##################################
# LDSC rg results
# ##################################
# Read bivariate LDSC regression results (with latest psychiatric results)
## LDSC files
ldscFiles=list.files("results/fastGWA/ldsc/", pattern="*.ldsc.rg.log")
# List of outcomes
outcome_info=read.xlsx("input/gsmr_traitList.xlsx")
outcome_info=outcome_info[outcome_info$Present_results=="Yes",]
outcomeList=outcome_info$File_prefix
ldscFiles=grep(paste(outcomeList,collapse="|"), ldscFiles, value=T)
# Table to keep ldsc rg results
ldscResults=data.frame(exposure=rep(c("vitD_fastGWA","vitD_BMIcov_fastGWA","vitD_BMIcond_fastGWA"), each=length(outcomeList)),
outcome=outcomeList,
outcomeName=NA, PMID=NA, Category=NA, h2=NA,
rg=NA, se=NA, p=NA, gcov_int=NA, gcov_int_se=NA)
## Fill table
for(i in 1:nrow(ldscResults))
{
exposure=ldscResults[i,"exposure"]
outcome=ldscResults[i,"outcome"]
# Read LDSC results
fileName=paste0("results/fastGWA/ldsc/",exposure,".",outcome,".ldsc.rg.log")
tmp=read.table(fileName, skip=30, fill=T, h=F, colClasses="character", sep="_")[,1]
# Add LDSC results
## Cross-trait LD Score regression intercept and standard error
x=strsplit(grep("Intercept",tmp, value=T)[3],":")[[1]][2]
gcov_int=strsplit(x,"\\(")[[1]][1]
gcov_int_se=sub(")","",strsplit(x,"\\(")[[1]][2])
## Genetic correlation and its SE
x=strsplit(grep("Genetic Correlation:",tmp, value=T),":")[[1]][2]
rg=strsplit(x,"\\(")[[1]][1]
se=gsub(")","",strsplit(x,"\\(")[[1]][2])
## P-value for rg
p=strsplit(grep("P: ",tmp,value=T),":")[[1]][2]
## Heritability of outcome trait
x=strsplit(grep("Total Observed scale h2:", tmp, value=T)[2],":")[[1]][2]
h2=strsplit(x,"\\(")[[1]][1]
## Add all above
ldscResults[i,c("rg","se","p","gcov_int","gcov_int_se","h2")]=c(rg, se, p, gcov_int, gcov_int_se, h2)
# Add information about the outcome GWAS
tmp=outcome_info[outcome_info$File_prefix==outcome,]
ldscResults[i,c("outcomeName","PMID","Category")]=tmp[,c("Trait","PMID","Category")]
}
# Format
numericCols=c("rg","se","p","gcov_int","gcov_int_se","h2")
ldscResults[,numericCols]=lapply(ldscResults[,numericCols], as.numeric)
n_outcomes=length(unique(ldscResults$outcome))
bonf_thresh=0.05/n_outcomes
ldscResults$signif=ifelse(ldscResults$p<0.05, "signif","non-signif")
ldscResults[!is.na(ldscResults$p) & ldscResults$p<bonf_thresh,"signif"]="multi-signif"
ldscResults$signif=factor(ldscResults$signif, levels=c("non-signif","signif","multi-signif"))
rgOrder=order(ldscResults[ldscResults$exposure=="vitD_fastGWA","rg"], decreasing=T)
ldscResults$outcomeName=factor(ldscResults$outcomeName, levels=ldscResults[ldscResults$exposure=="vitD_fastGWA","outcomeName"][rgOrder])
ldscResults$exposureName=factor(ldscResults$exposure,
levels=c("vitD_fastGWA","vitD_BMIcov_fastGWA","vitD_BMIcond_fastGWA"),
labels=c("25OHD\nNo ajustment for BMI","25OHD\nWith BMI covarite","25OHD\nConditioned on BMI (mtCOJO)"))
# Plot
# png("manuscript/submission2/Figures/figureS6_rg.png", width=9, height=6, units='in', bg="transparent", res=150)
ggplot(ldscResults, aes(x=outcomeName, y=rg, color=signif)) +
geom_point(size=1.5) +
coord_flip() +
geom_errorbar(aes(x=outcomeName, ymin=rg-1.96*se, ymax=rg+1.96*se), width=0, size=.9) +
facet_wrap(.~exposureName) +
geom_hline(yintercept=0) +
theme(panel.grid.major.y = element_blank()) +
labs(y="Genetic correlation (rg) and 95% CI", x="") +
scale_color_manual(values=c("grey", "#FAC477", "lightcoral"),
name="",
labels=c(expression('P'[xy]*'>0.05'),
expression('P'[xy]*'<0.05'),
bquote(P[xy]<~.(format(bonf_thresh,digits = 1)))),
drop=FALSE)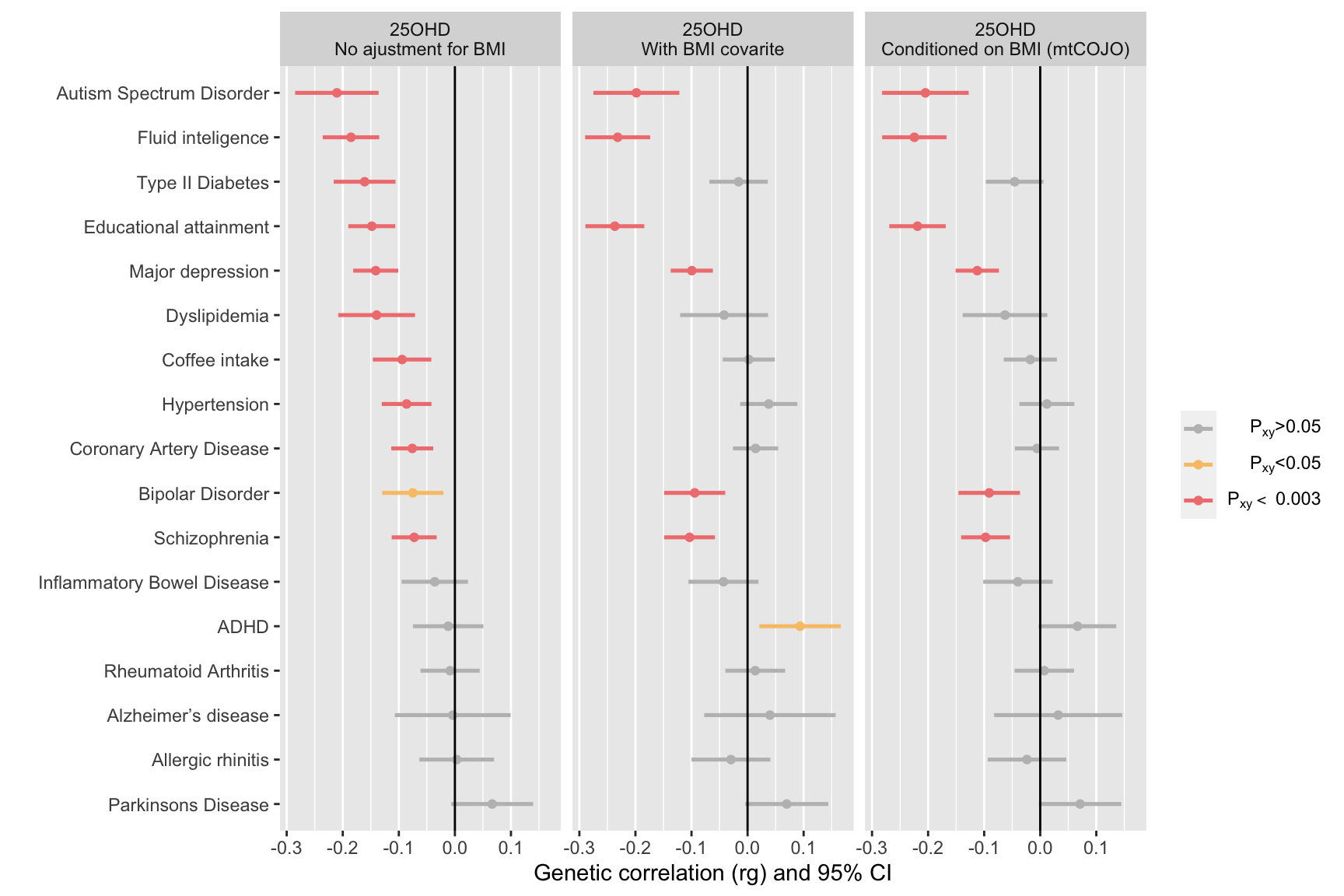
# dev.off()
# Format table (add LDHub results below)
ldscResults=ldscResults[,c("exposure","outcomeName","PMID","h2","rg","se","p")]
names(ldscResults)[1:2]=c("trait1","trait2")
ldscResults$LDHub="No"
# ##################################
# LD Hub rg results
# ##################################
# Merge results from vitD, vitD_BMIcov, and vitD_BMIcond with LDSC results above
## vitD
df=read.table("results/fastGWA/ldsc/vitD_fastGWA_withX.ldHubResults-rg.csv", sep=",", h=T, stringsAsFactors=F)
df=df[,c("trait1","trait2","PMID","h2_obs","rg","se","p")]
df$LDHub="Yes"
names(df)=names(ldscResults)
df$trait1="vitD_fastGWA"
## vitD_BMIcov
tmp=read.table("results/fastGWA/ldsc/vitD_BMIcov_fastGWA_withX.ldHubResults-rg.csv", sep=",", h=T, stringsAsFactors=F)
tmp=tmp[,c("trait1","trait2","PMID","h2_obs","rg","se","p")]
tmp$LDHub="Yes"
names(tmp)=names(df)
tmp$trait1="vitD_BMIcov_fastGWA"
df=rbind(df, tmp)
## vitD_BMIcond
tmp=read.table("results/fastGWA/ldsc/vitD_BMIcond_fastGWA.ldHubResults-rg.csv", sep=",", h=T, stringsAsFactors=F)
tmp=tmp[,c("trait1","trait2","PMID","h2_obs","rg","se","p")]
tmp$LDHub="Yes"
names(tmp)=names(df)
tmp$trait1="vitD_BMIcond_fastGWA"
df=rbind(df, tmp)
# Identify repeated studies with different results in LDHub and remove
df$flag=paste0(df$trait1,df$trait2,df$PMID)
df=df[!df$flag %in% df[duplicated(df$flag),"flag"],]
df$flag=NULL
# Merge tables and format
df=rbind(ldscResults, df)
## Round results
df$p=format(df$p, digits = 3)
df$se=round(df$se, 4)
## Remove traits with no results
df=df[!is.na(df$rg),]
# Change 25OHD trait labels
tmp=df
tmp$trait1=factor(tmp$trait1,
levels=c("vitD_fastGWA","vitD_BMIcov_fastGWA","vitD_BMIcond_fastGWA" ),
labels=c("25OHD - No ajustment for BMI","25OHD - With BMI covarite","25OHD - Conditioned on BMI (mtCOJO)" ))
# Present table
datatable(tmp, rownames=FALSE,
options=list(pageLength=5,
dom='frtipB',
buttons=c('csv', 'excel'),
scrollX=TRUE),
caption="Genetic correlation between vitamin D and LD Hub traits",
extensions=c('Buttons','FixedColumns')) # Table for publication (Rmd won't run dcast on multiple variables...)
if(F)
{
# Change 25OHD trait labels
df$trait1=factor(df$trait1,
levels=c("vitD_fastGWA","vitD_BMIcov_fastGWA","vitD_BMIcond_fastGWA" ),
labels=c("","BMIcov","BMIcond" ))
## Re-order columns for better comparisons
df=dcast(data.table::setDT(df), trait2 + PMID + LDHub ~ trait1, value.var=c("h2","rg","se","p"))
names(df)=sub("_$","",names(df))
names(df)[1]=""
## Order table by absolute rg
df=df[order(as.numeric(df$p)),]
## Save
write.xlsx(df, "manuscript/tables/tableS12_rg.xlsx", row.names=F)
}Columns are: trait1 and trait2, traits used to calculate genetic correlation, PMID, PubMed ID of trait2; h2, observed scale h2 for trait2 rg, se, and p, genetic correlation and respective standard error and p-value.
Reminder: If the rg estimate is above 1.25 or below -1.25, one of the
h2 estimates was very close to zero. Since h2 is
in the denominator of rg, if h2 is close to zero, rg
estimates can be very high.
GSMR
# Define traits to present results for
outcome_info=read.xlsx("input/gsmr_traitList.xlsx")
traits2exclude=outcome_info[outcome_info$Present_results=="No","Trait"]
traits2exclude=traits2exclude[!traits2exclude %in% outcome_info[outcome_info$Present_results=="Yes","Trait"]]
traits2exclude=gsub(" ","_", traits2exclude)
# GSMR results with no HEIDI filtering
tmp=read.table("results/fastGWA/gsmr/gsmr_with20kUKB_noHeidi.gsmr", h=T, stringsAsFactors=F)
tmp=tmp[!tmp$Exposure %in% traits2exclude & !tmp$Outcome %in% traits2exclude,]
n_outcomes=length(unique(tmp$Outcome))-3 # We ran bi-directional GSMR, so 3 outcome traits are vitD, vitD_BMIcov, and vitD_BMIcond
bonf_thresh=0.05/n_outcomes
tmp$signif=ifelse(tmp$p<0.05, "signif","non-signif")
tmp[!is.na(tmp$p) & tmp$p<bonf_thresh,"signif"]="multi-signif"
tmp$signif=factor(tmp$signif, levels=c("non-signif","signif","multi-signif"))
tmp$heidi="No HEIDI filetring"
gsmr=tmp
# GSMR results with HEIDI filtering
tmp=read.table("results/fastGWA/gsmr/gsmr_with20kUKB_withHeidi.gsmr", h=T, stringsAsFactors=F)
tmp=tmp[!tmp$Exposure %in% traits2exclude & !tmp$Outcome %in% traits2exclude,]
bonf_thresh=0.05/n_outcomes
tmp$signif=ifelse(tmp$p<0.05, "signif","non-signif")
tmp[!is.na(tmp$p) & tmp$p<bonf_thresh,"signif"]="multi-signif"
tmp$signif=factor(tmp$signif, levels=c("non-signif","signif","multi-signif"))
tmp$heidi="With HEIDI filetring"
gsmr=rbind(gsmr, tmp[,names(gsmr)])
# Vitamin D as exposure
tmp=gsmr[gsmr$Exposure %in% c("vitD","vitD_BMIcov","vitD_BMIcond"),]
bxyOrder=order(tmp[tmp$Exposure=="vitD" & tmp$heidi=="No HEIDI filetring","bxy"], decreasing=T)
tmp$Outcome=gsub("_"," ", tmp$Outcome)
ordered_phenos=tmp[tmp$Exposure=="vitD" & tmp$heidi=="No HEIDI filetring","Outcome"][bxyOrder]
tmp$Outcome=factor(tmp$Outcome, levels=ordered_phenos)
tmp$Exposure=factor(tmp$Exposure,
levels=c("vitD","vitD_BMIcov","vitD_BMIcond"),
labels=c("25OHD\nNo adjustment for BMI","25OHD\nWith BMI covarite","25OHD\nConditioned on BMI (mtCOJO)"))
#tmp$heidi=factor(tmp$heidi, levels=c("No HEIDI filetring","With HEIDI filetring"))
tmp$heidi=factor(tmp$heidi, levels=c("With HEIDI filetring","No HEIDI filetring"))
gsmr_vitD_exposure=tmp
# Vitamin D as outcome
tmp=gsmr[gsmr$Outcome %in% c("vitD","vitD_BMIcov","vitD_BMIcond"),]
tmp$Exposure=gsub("_"," ", tmp$Exposure)
bxyOrder=order(tmp[tmp$Outcome=="vitD" & tmp$heidi=="No HEIDI filetring","bxy"], decreasing=T)
tmp$Exposure=factor(tmp$Exposure, levels=ordered_phenos)
tmp$Outcome=factor(tmp$Outcome,
levels=c("vitD","vitD_BMIcov","vitD_BMIcond"),
labels=c("25OHD\nNo adjustment for BMI","25OHD\nWith BMI covarite","25OHD\nConditioned on BMI (mtCOJO)"))
#tmp$heidi=factor(tmp$heidi, levels=c("No HEIDI filetring","With HEIDI filetring"))
tmp$heidi=factor(tmp$heidi, levels=c("With HEIDI filetring","No HEIDI filetring"))
gsmr_vitD_outcome=tmpWe performed GSMR analyses between vitamin D and 17 outcome traits. The plot below depicts the estimated effect of vitamin D on the outcome traits. The nominal significance threshold used was 0.05. The significance threshold accounting for multiple testing was 0.0029412 (i.e. 0.05 / 17).
NB: We noticed, post-publication, that the GSMR results for ASD were mistakenly a duplication of the ADHD results in the plots below (Figure 5 in the paper). This error was fixed in this document and the effect size estimates of vitamin D on ASD remain small and largely non-significant, as presented in the manuscript. The number of SNP instruments were insufficient to assess the effect of ASD on vitamin D.
# Vitamin D as exposure
#tiff("manuscript/submission2/Figures/figure5a_gsmr.tiff", width=9, height=10, units='in', res=800,compression="lzw")
ggplot(gsmr_vitD_exposure, aes(x=Outcome, y=bxy, label=nsnp, color=signif)) +
geom_point(size=1.5) +
geom_text(vjust=-.5, aes(fontface=2), size=3)+
coord_flip() +
geom_errorbar(aes(x=Outcome, ymin=bxy-1.96*se, ymax=bxy+1.96*se), width=0, size=.8) +
facet_grid(heidi~Exposure) +
geom_hline(yintercept=0) +
theme(panel.grid.major.y = element_blank(),
plot.caption = element_text(hjust = 0)) +
labs(y="GSMR effect size (bxy) and 95% CI", x="",
#title="Effect of 25 hydroxyvitamin D on other traits",
title="(a) Effect of 25 hydroxyvitamin D on other traits",
caption="Numbers plotted represent the number of SNPs used in GSMR analyses") +
scale_color_manual(values=c("grey", "#FAC477", "lightcoral"),
name = "",
labels=c(expression('P'[xy]*'>0.05'),
expression('P'[xy]*'<0.05'),
bquote(P[xy]<~.(format(bonf_thresh,digits = 1)))),
drop = FALSE)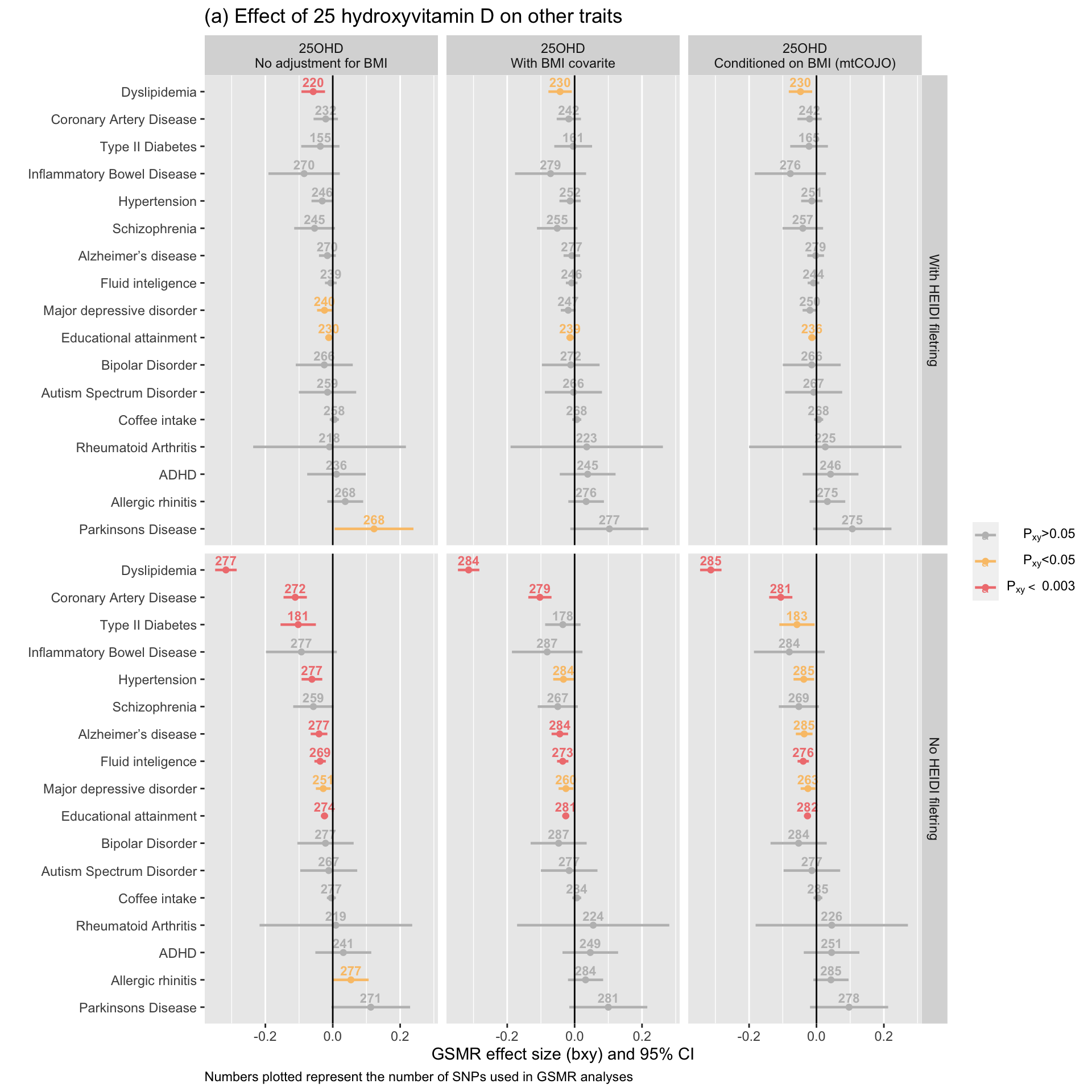
#dev.off()
# Vitamin D as outcome
#tiff("manuscript/submission2/Figures/figure5b_gsmr.tiff", width=9, height=10, units='in', res=800,compression="lzw")
ggplot(gsmr_vitD_outcome, aes(x=Exposure, y=bxy, label=nsnp, color=signif)) +
geom_point(size=1.5) +
geom_text(vjust=-.5, aes(fontface=2), size=3)+
coord_flip() +
geom_errorbar(aes(x=Exposure, ymin=bxy-1.96*se, ymax=bxy+1.96*se), width=0, size=.8) +
facet_grid(heidi~Outcome) +
geom_hline(yintercept=0) +
theme(panel.grid.major.y = element_blank(),
plot.caption = element_text(hjust = 0)) +
labs(y="GSMR effect size (bxy) and 95% CI", x="",
#title="Effect of other traits on 25 hydroxyvitamin D",
title="(b) Effect of other traits on 25 hydroxyvitamin D",
caption="Numbers plotted represent the number of SNPs used in GSMR analyses") +
scale_color_manual(values=c("grey", "#FAC477", "lightcoral"),
name = "",
labels=c(expression('P'[xy]*'>0.05'),
expression('P'[xy]*'<0.05'),
bquote(P[xy]<~.(format(bonf_thresh,digits = 1)))),
drop = FALSE)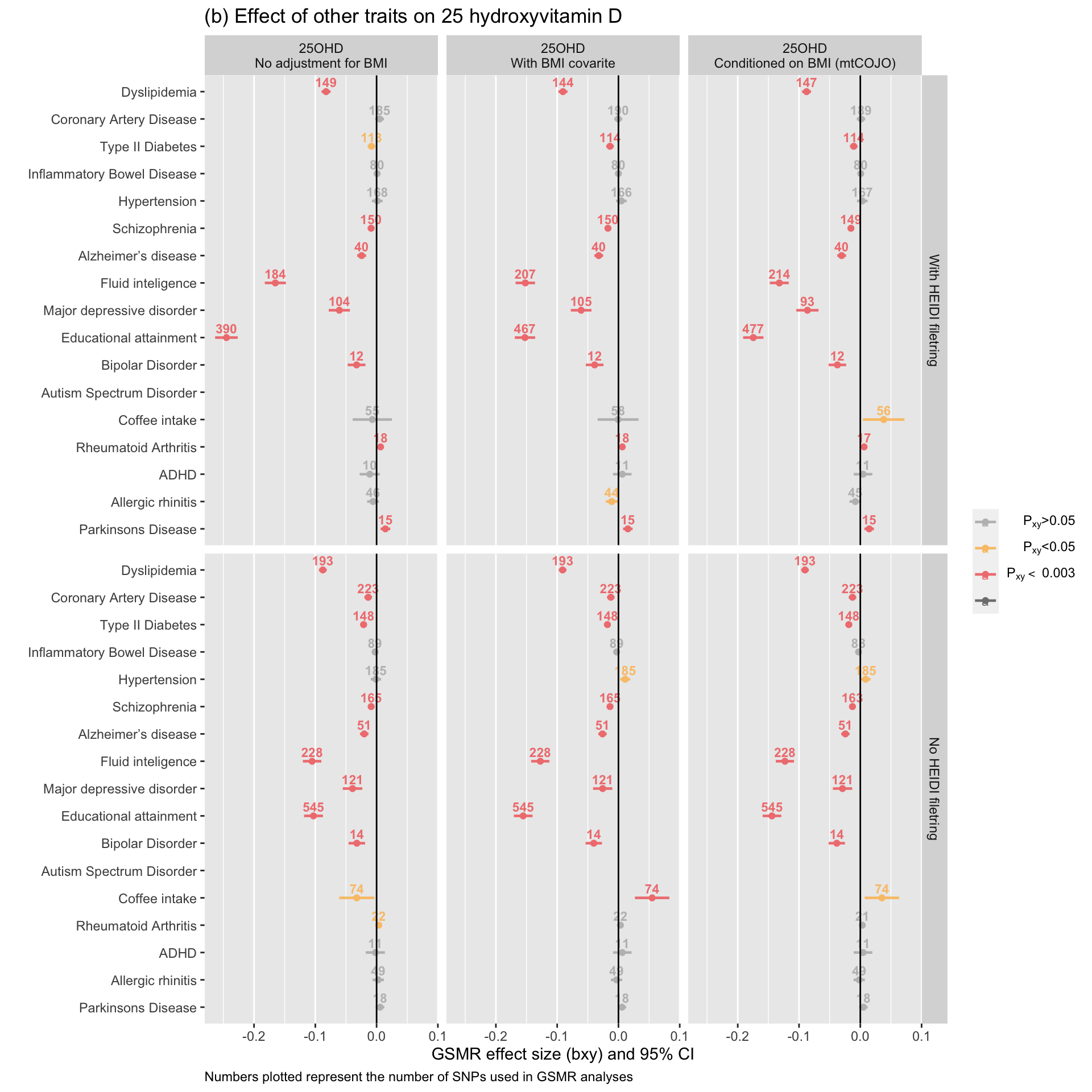
#dev.off()
# Present table
tmp=rbind(gsmr_vitD_exposure, gsmr_vitD_outcome)
tmp$Exposure=sub("\n"," - ",tmp$Exposure)
tmp$Outcome=sub("\n"," - ",tmp$Outcome)
tmp=tmp[order(tmp$p),c("Exposure","Outcome","heidi","bxy","se","p","nsnp")]
datatable(tmp, rownames=FALSE,
options=list(pageLength=5,
dom='frtipB',
buttons=c('csv', 'excel'),
scrollX=TRUE),
caption="Bi-directional GSMR associations between vitamin D and other traits",
extensions=c('Buttons','FixedColumns')) 2SMR
# Load and format 2SMR results
df=read.table("results/fastGWA/twosmr/all.2smr", h=T, stringsAsFactors=F, sep="\t")
df$heidi=gsub(".*\\.","",df$exposure)
df$exposure=gsub("\\..*","",df$exposure)
df=df[,c("exposure","outcome","method","heidi","nsnp","b","se","pval")]
## Round results
df$pval=format(df$pval, digits=3)
cols=grep("se|b",names(df)); df[,cols]=apply(df[,cols], 2, function(x){round(x, 4)})
# Present table
datatable(df, rownames=FALSE,
options=list(pageLength=5,
dom='frtipB',
buttons=c('csv', 'excel'),
scrollX=TRUE),
caption="2SMR results for causal associations identified with GSMR",
extensions=c('Buttons','FixedColumns')) %>%
formatStyle("pval","white-space"="nowrap")