25OHD phenotype
# Libraries
library(reshape2)
library(ggplot2)
library(knitr)
library(kableExtra)
library(data.table)
library(DT)
library(hrbrthemes)
# Functions
norm = function(x){x[order(x,na.last=NA)]=qnorm(ppoints(length(x[!is.na(x)])),lower=T);x }
se=function(x) {sqrt(var(x,na.rm=TRUE)/length(na.omit(x)))}
# Save results when running all chunks?
saveResults=F
saveplots=FVariables extracted
To generate phenotype files to use in the GWA analyses, we extracted the following data fields:
Data description
Overview
# Load tidied data
df=read.table("input/vitaminD_tidyData", h=T, stringsAsFactors=F)
df2=reshape2::dcast(df,IID~visit, value.var="vitaminD")
# Sample size with and without vitamin D levels assessed
tot=502536
vitD=nrow(df2)
f=round(vitD/tot*100,2)
# Sample sizes by instance
n1=nrow(df[df$visit=="v0",])
n2=nrow(df[df$visit=="v1",])
n02=nrow(df2[is.na(df2$v0) & !is.na(df2$v1),])
n12=length(unique(df[duplicated(df$IID),"IID"]))
# Restricted to EUR
v3EUR_HM3_IDs=read.table("input/UKB_v3EUR_HM3/ukbEUR_imp_chr1_v3_HM3.fam", h=F, stringsAsFactors=F)
nEUR=nrow(v3EUR_HM3_IDs)
n1_EUR=nrow(df[df$visit=="v0" & df$IID %in% v3EUR_HM3_IDs$V1,])
n02_EUR=nrow(df2[is.na(df2$v0) & !is.na(df2$v1) & df2$IID %in% v3EUR_HM3_IDs$V1,])
# Make table of sample sizes
tmp=data.frame(assessmentTime=c("First assessment at v0","First assessment at v1","Total"),
All=c(n1, n02, n1+n02),
Europeans=c(n1_EUR, n02_EUR, n1_EUR+n02_EUR))
names(tmp)[1]=""
tmp=kable(tmp, row.names=F)
kable_styling(tmp, full_width=F)
# Save env
vars2keep=c("f","n02_EUR","n1","n1_EUR","n12","n2","tmp","tot","vitD")
save(list=vars2keep, file = "rdm/input/rmd/dimensions.Rdata")We have data for 5.02536^{5} individuals. Of those, 449978 (89.54%) have vitamin D levels assessed:
- 448376 individuals assessed on v0 (2006-2010)
- 17039 individuals assessed on the v1 (2012-13)
15437 have vitamin D levels measured in both visits.
We restricted our analyses to the 1st vitamin D measurement that each individual has, and analysed individuals of European ancestry only (N=417580):
| All | Europeans | |
|---|---|---|
| First assessment at v0 | 448376 | 416072 |
| First assessment at v1 | 1602 | 1508 |
| Total | 449978 | 417580 |
Distribution
# Restrict analyses to 1st blood collection only (i.e. remove duplicate observations)
df=read.table("input/vitaminD_tidyData", h=T, stringsAsFactors=F)
df=df[df$analyse=="yes",]
df$analyse=NULL
# Restrict to individuals of European ancestry
df=df[df$IID %in% v3EUR_HM3_IDs$V1,]
# Summary of distribution
#se=function(x) {sqrt(var(x,na.rm=TRUE)/length(na.omit(x)))}
vitD=df$vitaminD
tmp=data.frame(t(unclass(summary(vitD))))
tmp$se=se(vitD); tmp$sd=sd(vitD,na.rm=T)
tmp$N=nrow(df)
tmp2=kable(tmp, caption=paste0("Vitamin D (nmol/L)"), row.names=F)
tmp2=kable_styling(tmp2, full_width=F)
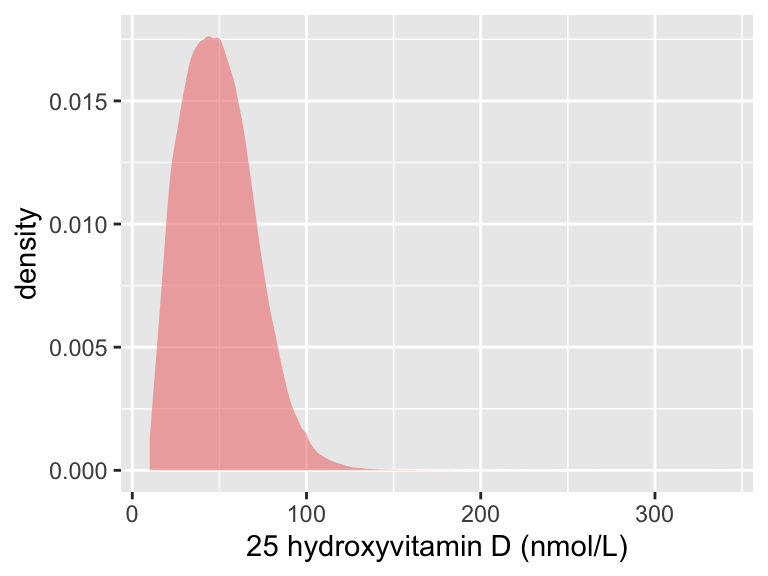
# Density plot of vitamin D
p1=ggplot(df, aes(x=vitaminD)) +
geom_density(fill="lightcoral", color=alpha("grey", 0),alpha = 0.6) +
labs(x="25 hydroxyvitamin D (nmol/L)")
# Save env
vars2keep=c("tmp2","p1")
save(list=vars2keep, file = "rdm/input/rmd/distribution.Rdata")
tmp2
p1| Min. | X1st.Qu. | Median | Mean | X3rd.Qu. | Max. | se | sd | N |
|---|---|---|---|---|---|---|---|---|
| 10 | 33.5 | 47.9 | 49.56348 | 63.2 | 340 | 0.0324702 | 20.9824 | 417580 |
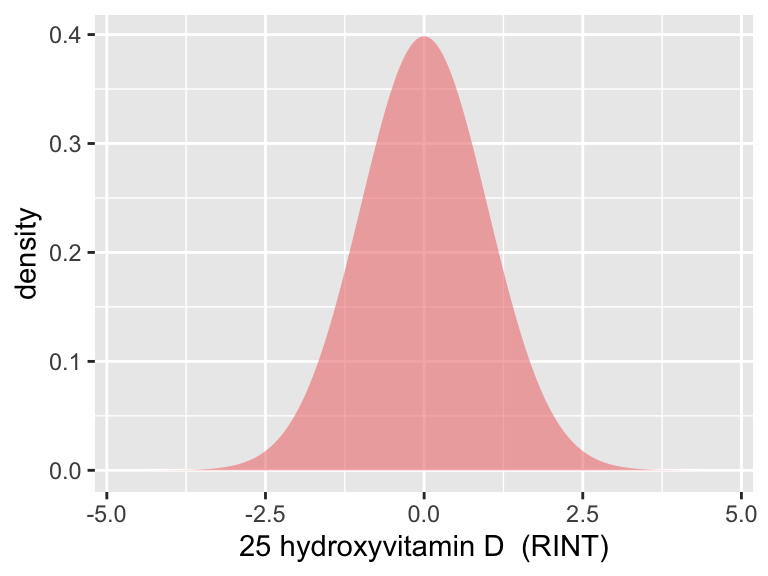
We applied a rank-based inverse-normal transformation to vitamin D levels.
# *************************
# Normalise vitamin D
# *************************
#norm = function(x){x[order(x,na.last=NA)]=qnorm(ppoints(length(x[!is.na(x)])),lower=T);x }
df$norm_vitD=norm(df$vitaminD)
df$log_vitD=log(df$vitaminD)
## Save for all descriptive analayses below
df$season=NULL
if(saveResults){write.table(df, "input/vitaminD_tidyData_noMissing", quote=F, row.names=F)}
# Summary of normalised distribution
df=read.table("input/vitaminD_tidyData_noMissing", h=T, stringsAsFactors=F)
vitD=df$norm_vitD
tmp=data.frame(t(unclass(summary(vitD))))
tmp$se=se(vitD); tmp$sd=sd(vitD,na.rm=T)
tmp$N=nrow(df)
tmp2=kable(tmp, caption=paste0("Vitamin D (RINT)"), row.names=F)
tmp2=kable_styling(tmp2, full_width=F)
# Density plot of normalised vitamin D
p1=ggplot(df, aes(x=norm_vitD)) +
geom_density(fill="lightcoral", color=alpha("grey", 0),alpha = 0.6) +
labs(x="25 hydroxyvitamin D (RINT)")
# Save env
vars2keep=c("tmp2","p1")
save(list=vars2keep, file = "rdm/input/rmd/nomalised_distribution.Rdata")
tmp2
p1| Min. | X1st.Qu. | Median | Mean | X3rd.Qu. | Max. | se | sd | N |
|---|---|---|---|---|---|---|---|---|
| -4.716892 | -0.6744879 | 0 | 0 | 0.6744879 | 4.716892 | 0.0015475 | 0.9999996 | 417580 |
Time series
Vitamin D in Year-Months
df=read.table("input/vitaminD_tidyData_noMissing", h=T, stringsAsFactors=F)
# *************************
# Average
# *************************
# Calculate average and 95% CI in each month
df$assessMonthYear=substr(df$assessDate,1,7)
df_YMmeans = do.call(data.frame,aggregate(norm_vitD ~ assessMonthYear+visit, df, function(x)c(mean=mean(x), sd=sd(x), N=length(x))))
names(df_YMmeans)[3:5] = c("norm_vitD","sd","N")
df_YMmeans$minCI = df_YMmeans$norm_vitD - 1.96*df_YMmeans$sd/sqrt(df_YMmeans$N)
df_YMmeans$maxCI = df_YMmeans$norm_vitD + 1.96*df_YMmeans$sd/sqrt(df_YMmeans$N)
# Sample size per month
tmp=data.frame(table(df$assessMonthYear, df$visit))
tmp=tmp[tmp$Freq!=0,]
names(tmp)=c("ym","visit","freq")
# Remove outliers
outliers=head(tmp[order(tmp$freq),])
df_YMmeans=df_YMmeans[!df_YMmeans$assessMonthYear %in% outliers[outliers$freq<30,"ym"],]
# Change visit labels
df_YMmeans$visit=factor(df_YMmeans$visit, labels=c("Baseline", "Follow-up"))
# Plot monthly average
p1=ggplot(df_YMmeans, aes(x=assessMonthYear, y=norm_vitD, group=visit)) +
geom_ribbon(aes(ymin=minCI,ymax=maxCI),alpha=0.3, color=NA, fill="lightcoral")+
geom_line(size=1.1, col="lightcoral") +
geom_point(size=4, aes(shape=visit), fill="white", col="lightcoral") +
scale_shape_manual(values = c(22, 24)) +
theme(text = element_text(size=13),
axis.text.x = element_text(angle = 45, vjust = 1, hjust=1),
legend.title = element_blank(),
legend.position = "none",
plot.background = element_rect(fill="transparent",colour = NA)) +
labs(y="Average 25 hydroxyvitamin D\n(RINT)", x=paste0("Month and Year of assessment")) +
theme(plot.caption = element_text(hjust = 0))
# Save env
vars2keep=c("tmp","p1")
save(list=vars2keep, file = "rdm/input/rmd/vitD_ym.RData")
if(saveplots){tiff("manuscript/Figures/figureS1_25OHD_mean_RINT_YM.tiff", width=10, height=4 ,units="in", res=800,compression = "lzw", bg="transparent")}
p1
if(saveplots){dev.off()}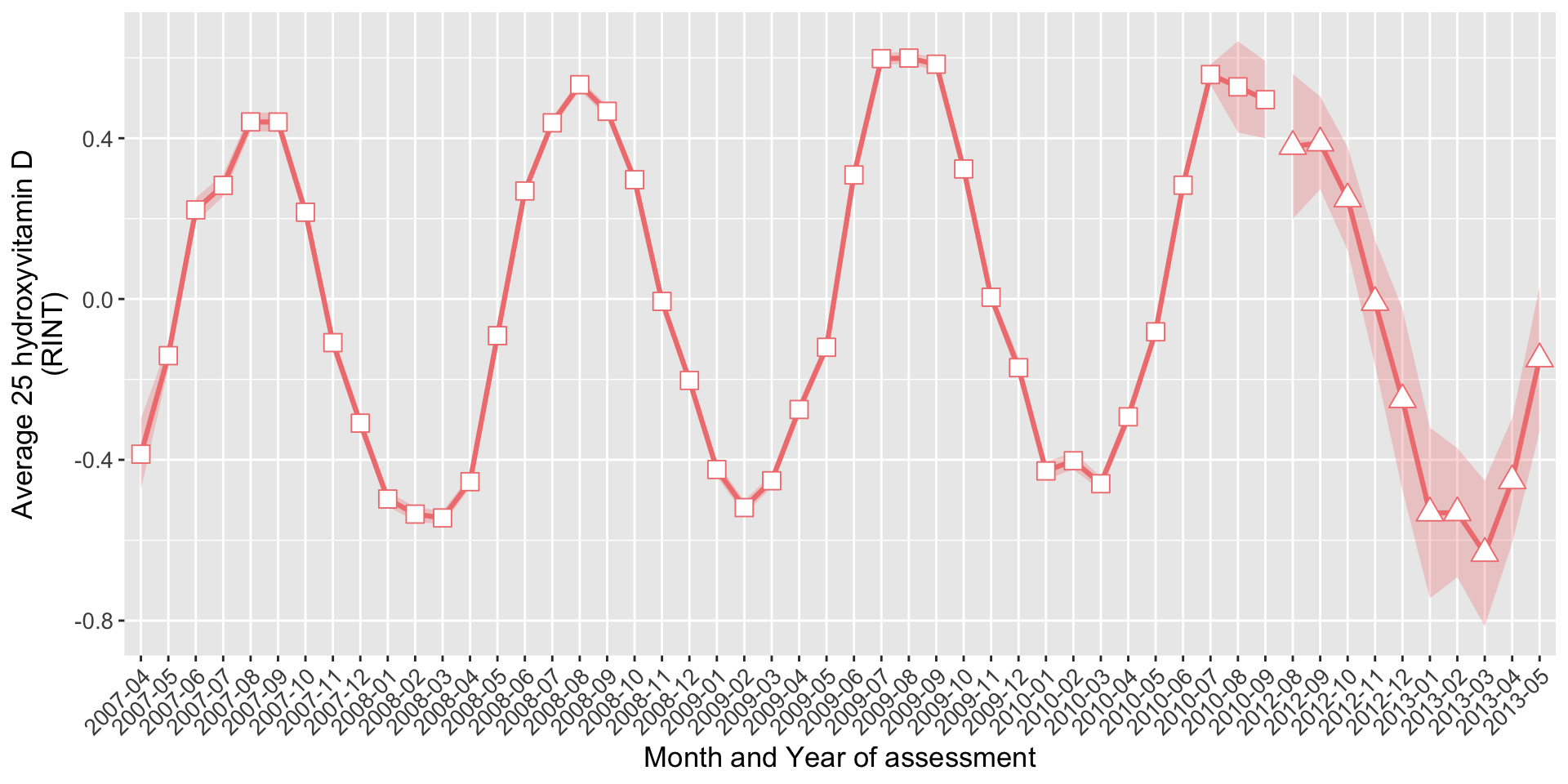 On average:
On average:
- 9456 individuals were assessed per month in the first visit (v0)
- 137 individuals were assessed per month in the repeat visit (v1)
Vitamin D in each month (all years aggregated)
df=read.table("input/vitaminD_tidyData_noMissing", h=T, stringsAsFactors=F)
# *************************
# Average
# *************************
# ----- Monthly mean of 25OHD (obs scale)
# Calculate average and 95% CI in each month
tmp = do.call(data.frame,aggregate(vitaminD ~ assessMonth, df, function(x)c(mean=mean(x), sd=sd(x), N=length(x))))
names(tmp)[2:4] = c("vitD","sd","N")
tmp[tmp$assessMonth>=12 | tmp$assessMonth<=4,"season"]="winter"
tmp[tmp$assessMonth>=6 & tmp$assessMonth<=10,"season"]="summer"
tmp[tmp$assessMonth==5 | tmp$assessMonth==11,"season"]="N/A"
tmp$season=factor(tmp$season, levels=c("summer","winter","N/A"))
tmp$minCI = tmp$vitD - 1.96*tmp$sd/sqrt(tmp$N)
tmp$maxCI = tmp$vitD + 1.96*tmp$sd/sqrt(tmp$N)
# Plot
p1=ggplot(data=tmp, aes(x=assessMonth, y=vitD, group=1)) +
geom_ribbon(data=tmp,aes(ymin=minCI,ymax=maxCI), alpha=0.4, color=NA, fill="grey") +
geom_line(size=1, col="grey") +
geom_point(size=5, aes(col=season)) +
scale_color_manual(values=c("orange","cadetblue","grey")) +
theme(legend.title = element_blank(),
legend.position = "none",
plot.background = element_rect(fill="transparent",colour = NA)) +
scale_x_continuous(breaks=seq(1,12,1)) +
labs(y="Average 25 hydroxyvitamin D (nmol/L)", x=paste0("Month of assessment"))
if(saveplots){tiff("manuscript/Figures/figureS1_25OHD_mean_obsScale.tiff", width=5, height=4 ,units="in", res=800,compression = "lzw", bg="transparent")}
p1
if(saveplots){dev.off()}
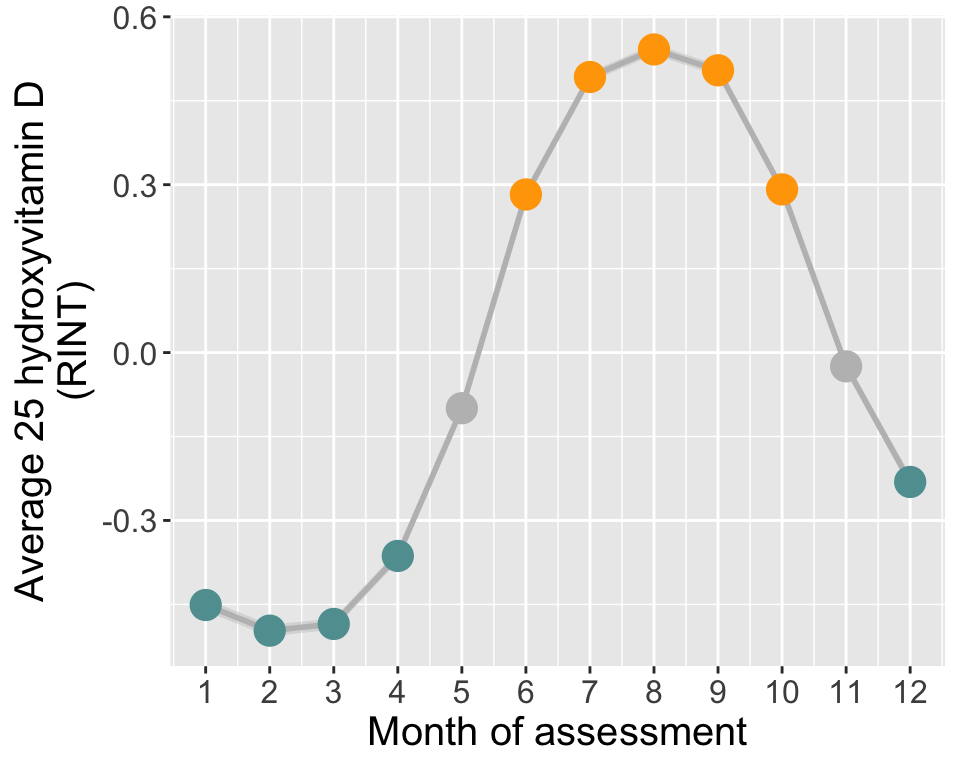
# ----- Monthly mean of 25OHD (RINT)
# Calculate average and 95% CI in each month
tmp = do.call(data.frame,aggregate(norm_vitD ~ assessMonth, df, function(x)c(mean=mean(x), sd=sd(x), N=length(x))))
names(tmp)[2:4] = c("vitD","sd","N")
tmp[tmp$assessMonth>=12 | tmp$assessMonth<=4,"season"]="winter"
tmp[tmp$assessMonth>=6 & tmp$assessMonth<=10,"season"]="summer"
tmp[tmp$assessMonth==5 | tmp$assessMonth==11,"season"]="N/A"
tmp$season=factor(tmp$season, levels=c("summer","winter","N/A"))
tmp$minCI = tmp$vitD - 1.96*tmp$sd/sqrt(tmp$N)
tmp$maxCI = tmp$vitD + 1.96*tmp$sd/sqrt(tmp$N)
# Plot
p2=ggplot(data=tmp, aes(x=assessMonth, y=vitD, group=1)) +
geom_ribbon(data=tmp,aes(ymin=minCI,ymax=maxCI), alpha=0.4, color=NA, fill="grey") +
geom_line(size=1, col="grey") +
geom_point(size=5, aes(col=season)) +
scale_color_manual(values=c("orange","cadetblue","grey")) +
theme(text = element_text(size=15),
legend.title = element_blank(),
legend.position = "none",
plot.background = element_rect(fill="transparent",colour = NA)) +
scale_x_continuous(breaks=seq(1,12,1)) +
labs(y="Average 25 hydroxyvitamin D\n(RINT)", x=paste0("Month of assessment"))
if(saveplots){tiff("manuscript/Figures/figureS1_25OHD_mean_RINT.tiff", width=5, height=4 ,units="in", res=800,compression = "lzw", bg="transparent")}
p2
if(saveplots){dev.off()}
# *************************
# Variance
# *************************
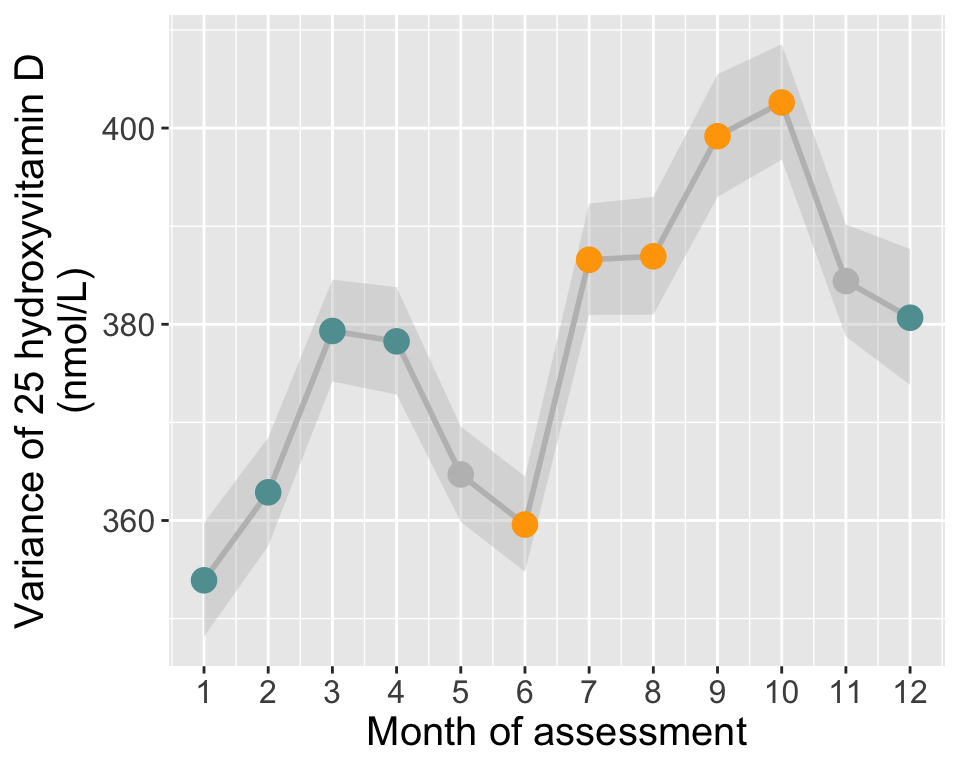
# Calculate variance and 95% CI in each month
tmp = do.call(data.frame,aggregate(vitaminD ~ assessMonth, df, function(x)c(var=var(x), N=length(x))))
names(tmp)[2:3] = c("vitD_s2","N")
tmp[tmp$assessMonth>=12 | tmp$assessMonth<=4,"season"]="winter"
tmp[tmp$assessMonth>=6 & tmp$assessMonth<=10,"season"]="summer"
tmp[tmp$assessMonth==5 | tmp$assessMonth==11,"season"]="N/A"
tmp$season=factor(tmp$season, levels=c("summer","winter","N/A"))
chi2_0.025=qchisq(0.025,tmp$N)
chi2_0.975=qchisq(0.975,tmp$N)
tmp$minCI = (tmp$N-1)*tmp$vitD_s2/chi2_0.975
tmp$maxCI = (tmp$N-1)*tmp$vitD_s2/chi2_0.025
# Plot
p3=ggplot(data=tmp, aes(x=assessMonth, y=vitD_s2, group=1)) +
geom_ribbon(data=tmp,aes(ymin=minCI,ymax=maxCI),alpha=0.4, color=NA, fill="grey")+
geom_line(size=1, color="grey") +
geom_point(size=4, aes(col=season)) +
scale_color_manual(values=c("orange","cadetblue","grey")) +
scale_shape_manual(values = c(21, 24)) +
theme(text = element_text(size=15),
legend.title = element_blank(),
legend.position = "none",
plot.background = element_rect(fill="transparent",colour = NA)) +
scale_x_continuous(breaks=seq(1,12,1)) +
labs(y="Variance of 25 hydroxyvitamin D\n(nmol/L)", x=paste0("Month of assessment"))
if(saveplots){tiff("manuscript/Figures/figureS1_25OHD_variance_obsScale.tiff", width=5, height=4 ,units="in", res=800,compression = "lzw", bg="transparent")}
p3
if(saveplots){dev.off()}
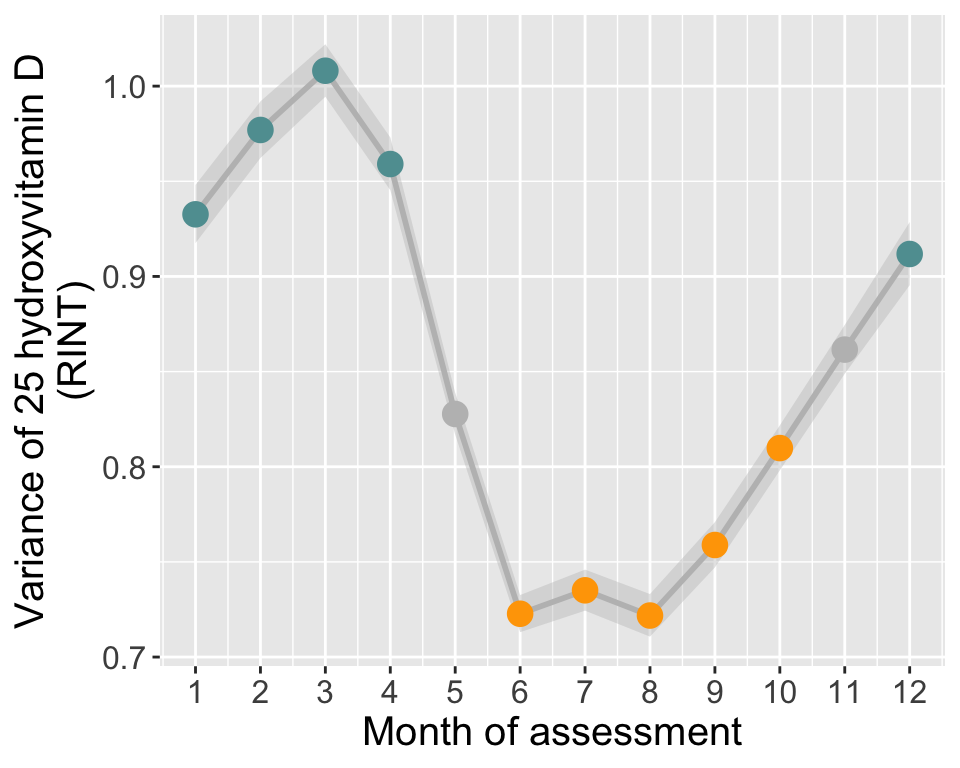
# ----- Monthly variance of 25OHD (RINT)
# Calculate variance and 95% CI in each month
tmp = do.call(data.frame,aggregate(norm_vitD ~ assessMonth, df, function(x)c(var=var(x), N=length(x))))
names(tmp)[2:3] = c("vitD_s2","N")
tmp[tmp$assessMonth>=12 | tmp$assessMonth<=4,"season"]="winter"
tmp[tmp$assessMonth>=6 & tmp$assessMonth<=10,"season"]="summer"
tmp[tmp$assessMonth==5 | tmp$assessMonth==11,"season"]="N/A"
tmp$season=factor(tmp$season, levels=c("summer","winter","N/A"))
chi2_0.025=qchisq(0.025,tmp$N)
chi2_0.975=qchisq(0.975,tmp$N)
tmp$minCI = (tmp$N-1)*tmp$vitD_s2/chi2_0.975
tmp$maxCI = (tmp$N-1)*tmp$vitD_s2/chi2_0.025
# Plot
p4=ggplot(data=tmp, aes(x=assessMonth, y=vitD_s2, group=1)) +
geom_ribbon(data=tmp,aes(ymin=minCI,ymax=maxCI),alpha=0.4, color=NA, fill="grey")+
geom_line(size=1, color="grey") +
geom_point(size=4, aes(col=season)) +
scale_color_manual(values=c("orange","cadetblue","grey")) +
scale_shape_manual(values = c(21, 24)) +
theme(text = element_text(size=15),
legend.title = element_blank(),
legend.position = "none",
plot.background = element_rect(fill="transparent",colour = NA)) +
scale_x_continuous(breaks=seq(1,12,1)) +
labs(y="Variance of 25 hydroxyvitamin D\n(RINT)", x=paste0("Month of assessment"))
if(saveplots){tiff("manuscript/Figures/figureS1_25OHD_variance_RINT.tiff", width=5, height=4 ,units="in", res=800,compression = "lzw", bg="transparent")}
p4
if(saveplots){dev.off()}
# Save env
vars2keep=c("p1","p2","p3","p4")
save(list=vars2keep, file = "rdm/input/rmd/vitD_monthly_mean_and_var.RData")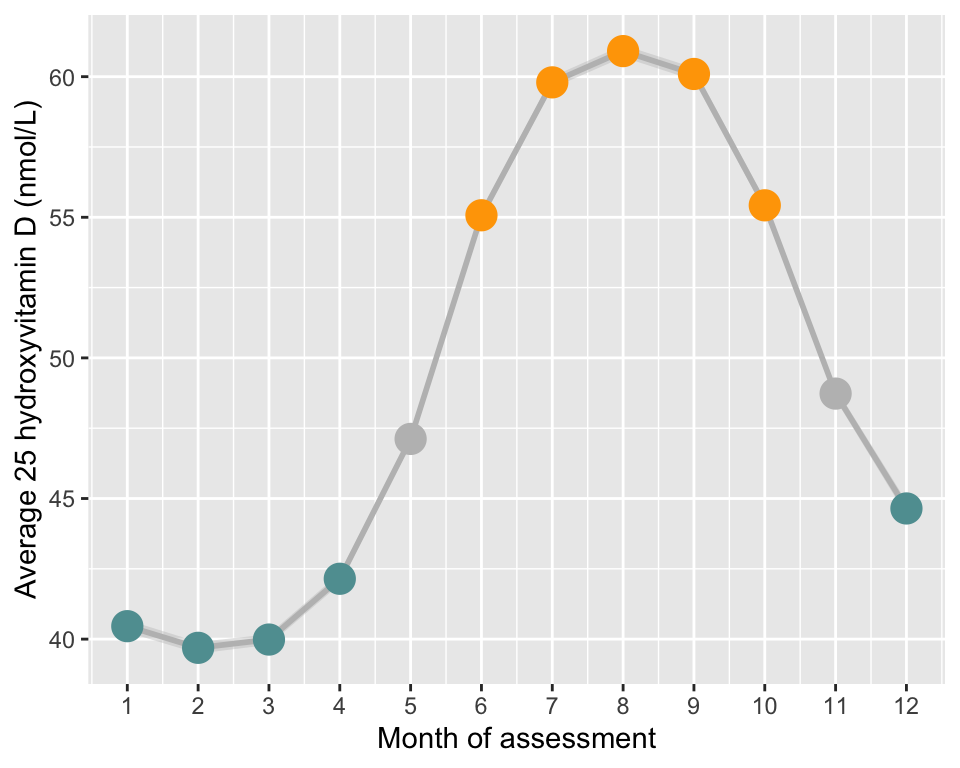
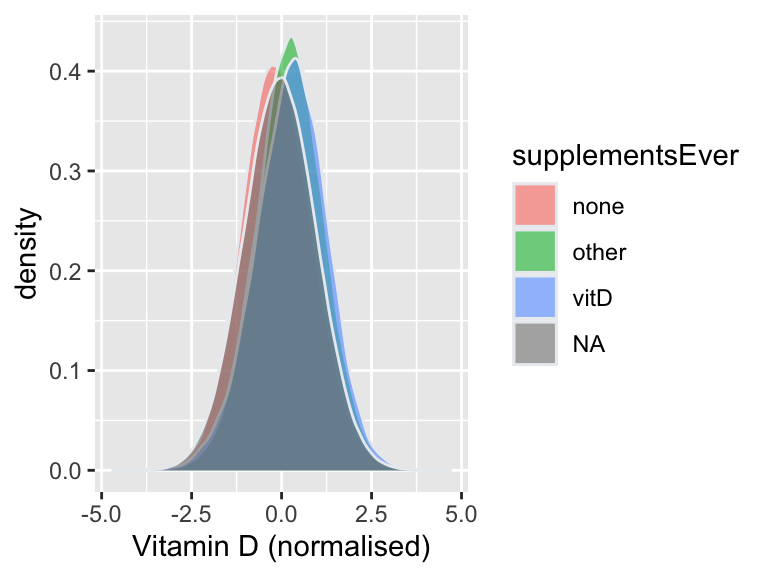
Vitamin D supplements
Field 104670 indicates whether individuals replied to the survey on supplement intake.
For individuals who replied yes, field 20084 summaries which supplements they took. Individuals with code 479 in that question indicated that they had vitamin D supplements the day before the questionnaire.
Fields 104670 and 20084 were assessed at 5 instances:
- instance 0: April 2009 to September 2010
- instance 1: February 2011 to April 2011
- instance 2: June 2011 to September 2011
- instance 3: October 2011 to December 2011
- instance 4: April 2012 to June 2012
Vitamin D in individuals with/without supplements at any time point
Using information from all time-points, the following groups were defined:
- NA: Individuals with no information on supplements taken (ever)
- none: Individuals who always reported not taking supplements
- other: Individuals who always reported taking other supplements (not vitamin D)
- vitD: Individuals who ever reported taking vitamin D (may also have taken other supplements)
As before, the group no-vitD was obtained by grouping individuals in the categories none and other.
df=read.table("input/vitaminD_tidyData_noMissing", h=T, stringsAsFactors=F)
# Summary in individuals with no supplement information
vitD=df[is.na(df$supplementsEver),"norm_vitD"]
n=length(vitD)
tmp=data.frame(t(unclass(summary(vitD))))
tmp$se=se(vitD); tmp$sd=sd(vitD,na.rm=T); tmp$N=n
tmp$supplementGroup="NA"
df2=tmp[,c(ncol(tmp), ncol(tmp)-1,1:(ncol(tmp)-2))]
# Summary in individuals with supplement information
for(i in c("none","other","vitD"))
{
vitD=df[!is.na(df$supplementsEver) & df$supplementsEver==i,"norm_vitD"]
n=length(vitD)
tmp=data.frame(t(unclass(summary(vitD))))
tmp$se=se(vitD); tmp$sd=sd(vitD,na.rm=T); tmp$N=n
tmp$supplementGroup=i
df2=rbind(df2, tmp[,names(df2)])
}
# Summary in individuals with no vitD supplements (i.e. 'none'+'other')
vitD=df[!is.na(df$supplementsEver) & df$supplementsEver %in% c("none","other"),"norm_vitD"]
n=length(vitD)
tmp=data.frame(t(unclass(summary(vitD))))
tmp$se=se(vitD); tmp$sd=sd(vitD,na.rm=T); tmp$N=n
tmp$supplementGroup="no-vitD"
df2=rbind(df2, tmp[,names(df2)])
# Summary in individuals with ever vitamin D vs. never/NA
df$supplementGroup=ifelse(!is.na(df$supplements) & df$supplements=="vitD", "ever", "never/NA")
vitD=df[df$supplementGroup=="never/NA","norm_vitD"]
n=length(vitD)
tmp=data.frame(t(unclass(summary(vitD))))
tmp$se=se(vitD); tmp$sd=sd(vitD,na.rm=T); tmp$N=n
tmp$supplementGroup="never/NA (none+other+NA)"
df2=rbind(df2, tmp[,names(df2)])
# Summary table for all groups above
tmp2=kable(df2, caption=paste0("Vitamin D (normalised) by supplement group - EVER"), row.names=F)
tmp2=kable_styling(tmp2, full_width=F)
# Density plot by supplement group
p1=ggplot(df, aes(x=norm_vitD)) +
geom_density(aes(fill=supplementsEver), color="#e9ecef",alpha = 0.6) +
xlab("Vitamin D (normalised)")
# Density plot by supplement group: ever vs. never/NA
p2=ggplot(df, aes(x=norm_vitD)) +
geom_density(aes(fill=supplementGroup), color="#e9ecef",alpha = 0.6) +
xlab("Vitamin D (normalised)")
# Save env
vars2keep=c("p1","p2","tmp2")
save(list=vars2keep, file = "rdm/input/rmd/dist_by_supp.RData")| supplementGroup | N | Min. | X1st.Qu. | Median | Mean | X3rd.Qu. | Max. | se | sd |
|---|---|---|---|---|---|---|---|---|---|
| NA | 238090 | -4.716892 | -0.7236918 | -0.0414398 | -0.0407201 | 0.6418865 | 4.378065 | 0.0020718 | 1.0109343 |
| none | 90992 | -4.304144 | -0.7914820 | -0.1441555 | -0.1211890 | 0.5305524 | 4.716892 | 0.0032528 | 0.9812146 |
| other | 76863 | -3.947332 | -0.3846385 | 0.2374143 | 0.2283545 | 0.8575268 | 4.248166 | 0.0034084 | 0.9449495 |
| vitD | 11635 | -3.733036 | -0.3782312 | 0.3038705 | 0.2724769 | 0.9492897 | 4.132261 | 0.0091830 | 0.9905259 |
| no-vitD | 167855 | -4.304144 | -0.6208657 | 0.0357630 | 0.0388716 | 0.6970480 | 4.716892 | 0.0023929 | 0.9803668 |
| never/NA (none+other+NA) | 415693 | -4.716892 | -0.6772090 | -0.0031484 | -0.0029033 | 0.6712566 | 4.716892 | 0.0015505 | 0.9996895 |
In the plot below, the following groups were used:
- ever: vitamin D reported at any of the 5 time
points (may also have taken other supplements). Equivalent to
vitDabove. - never/NA: individuals who (1) never reported taking
vitamin D (may have reported other supplements or not taking supplements
ever) or (2) never replied to the supplement questionnaire. This is
equivalent to
other+none+NA.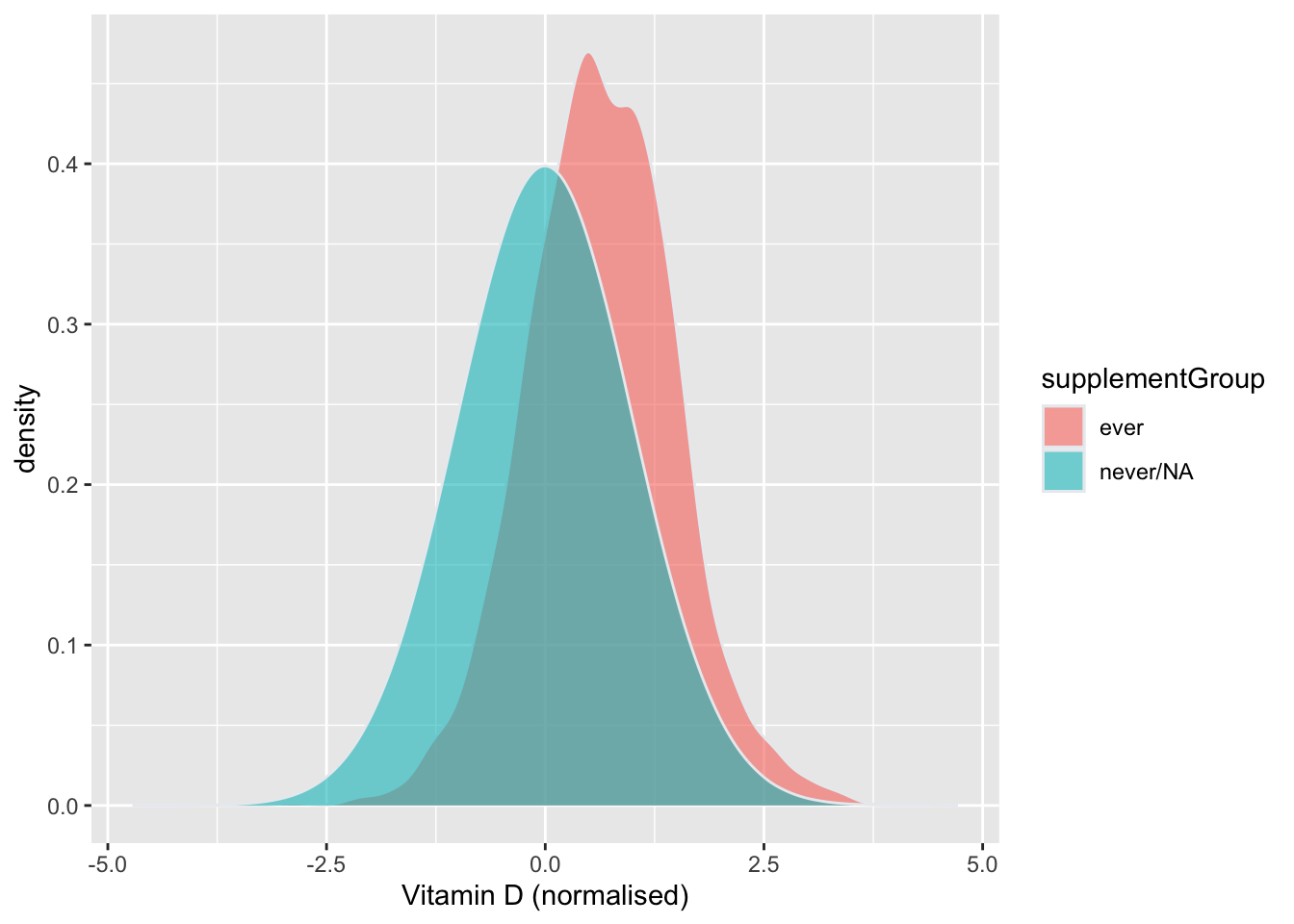
Covariates
Principal components
We included principal components (PCs) in our GWAS to account for population stratification. Specifically, we used 40 PCs generated with FlashPCA, using ~500K HapMap3 SNPs and excluding regions of long-range LD (LD<0.9).
Association tests with covariates
In this section we tested the association between potential confounder variables and normalised vitamin D levels.
df=read.table("input/vitaminD_tidyData_noMissing", h=T, stringsAsFactors=F)
df$assessMonthYear=substr(df$assessDate,1,7)
load("input/ukbEUR_PC40_HM3excludeRegions_v2.RData")
# Create file with phenotype and covariates to analyse
df2=df[,c("IID","vitaminD","norm_vitD","log_vitD","sex","age","bmi","batch","assessCentre","assessMonth","assessMonthYear","supplementsEver")]
df2=merge(df2, pcs[,2:12], all.x=T)
catVars=c("assessCentre","assessMonth","assessMonthYear","batch","supplementsEver")
df2[,catVars] = lapply(df2[,catVars], factor)
# Create a table to keep results from all association tests
out=data.frame(covariate=names(df2)[-c(1:4)],
test="lm",beta=NA, r2=NA, p=NA, stringsAsFactors=F)
out[out$covariate %in% catVars,"test"]="anova"
out2=c()
for(iv in c("vitaminD","norm_vitD","log_vitD"))
{
tmp=out
tmp$IV=iv
out2=rbind(out2,tmp)
}
out=out2
# Run associations
for (i in 1:nrow(out))
{
iv=out[i,"IV"]
c=out[i,"covariate"]
if(!c %in% catVars)
{
model=lm(df2[,iv]~df2[,c])
r2=summary(model)$r.squared
coef=summary(model)$coefficients
beta=round(coef[2,1],3)
p=coef[2,4]
}
if(c %in% catVars)
{
model=lm(df2[,iv]~df2[,c])
r2=summary(model)$r.squared
coef=summary(aov(df2[,iv]~df2[,c]))
beta=NA
p=coef[[1]][["Pr(>F)"]][1]
}
out[i,c("beta","r2","p")]=c(beta,
format(r2, digits=2),
format(p, digits=3))
}
# Run a few extra associations
if(F)
{
df2=merge(df2, pcs[,c(2,13:42)], all.x=T)
lmp <- function (modelobject) {
if (class(modelobject) != "lm") stop("Not an object of class 'lm' ")
f <- summary(modelobject)$fstatistic
p <- pf(f[1],f[2],f[3],lower.tail=F)
attributes(p) <- NULL
return(p)}
tmp=c()
# All 40 PCs
for(iv in c("vitaminD","norm_vitD","log_vitD"))
{
fixed=paste(paste0("PC",1:40), collapse="+")
model = as.formula(paste("df[,",iv,"]~",fixed))
test=lm(model, data=df2)
r2=summary(test)$r.squared
p=lmp(test)
tmp=data.frame(covariate="First 40 PCs", test="lm", beta=NA, r2=r2, p=p, IV=iv)
}
}
# Show results
out$p=format(out$p, digits=3)
out$p=as.character(out$p)
out=dcast(setDT(out), covariate +test ~ IV, value.var=c("beta","r2","p"))
# Save env
save(out, file = "rdm/input/rmd/cov_associations.RData")
datatable(out,
rownames=FALSE,
extensions=c('Buttons','FixedColumns'),
options=list(dom='frtipB',
buttons=c('csv', 'excel'),
scrollX=TRUE),
caption="Covariate association results with vitamin D (raw, RINT, and log-transformed)") Prepare file with covariates to include in MLM GWAS
We fit the following variables in the mixed linear model (MLM) analyses:
- Age
- Sex
- Assessment month
- Assessment centre
- First 40 PCs
- Genotyping batch
- Supplement intake: As defined above.
df=read.table("input/vitaminD_tidyData_noMissing", h=T, stringsAsFactors=F)
#load("input/ukbEUR_PC40_HM3excludeRegions_v2.RData")
# ---- Categorical confounders
df$FID=df$IID
catCov=df[,c("FID","IID","sex","batch","assessCentre","assessMonth","supplementsEver")]
catCov[is.na(catCov$supplementsEver),"supplementsEver"]="NoInfo"
# Create file with no sex for X chromosome analyses (which are run individually in males and females)
catCov_noSex=catCov[,grep("sex",names(catCov), invert=T)]
# ---- Quantitative confounders
quantCov=df[,c("FID","IID","age")]
quantCov=merge(quantCov, pcs[,2:42], all.x=T)
names(quantCov)[1:2]=c("FID","IID")
# Create a file with BMI as well
quantCov_bmi=df[,c("FID","IID","age","bmi")]
quantCov_bmi=merge(quantCov_bmi, pcs[,2:42], all.x=T)
names(quantCov_bmi)[1:2]=c("FID","IID")
# ---- Save files
if(saveResults){write.table(catCov, "input/categoricalCovariates", quote=F, row.names=F)}
if(saveResults){write.table(catCov_noSex, "input/categoricalCovariates_noSex", quote=F, row.names=F)}
if(saveResults){write.table(quantCov, "input/quantitativeCovariates", quote=F, row.names=F)}
if(saveResults){write.table(quantCov_bmi, "input/quantitativeCovariates_bmi", quote=F, row.names=F)}Prepare phenotype
In this section, we prepared the phenotypes for the following analyses:
fastGWA
The main GWA analyses were conducted with the mixed linear model (MLM) implemented in fastGWA
To generate the phenotype for those analyses, we:
- Restricted our sample to the full set of individuals with European ancestry
- Normalised phenotype with a rank-based inverse normal transformation (RINT)
Covariates were included in the GWA model to correct for confounder
effects. The list of confouders used is in the Covariate
section
# Restrict to ALL individuals of European ancestry
v3EUR_HM3_IDs=read.table("input/UKB_v3EUR_HM3/ukbEUR_imp_chr1_v3_HM3.fam", h=F, stringsAsFactors=F)
names(v3EUR_HM3_IDs)[1:2]=c("FID","IID")
df=read.table("input/vitaminD_tidyData_noMissing", h=T, stringsAsFactors=F)
df2=df[df$IID %in% v3EUR_HM3_IDs$IID,]
# Save log-transformed and RINT phenotype (to assess validity of meta-analysis)
if(saveResults){write.table(df2[,c("IID","log_vitD","norm_vitD")], "tmpDir/mlm_norm_log_pheno", row.names=F, quote=F)}
# Save normalised residuals to feed as phenotype to the fastGWA and BOLT-LMM
df2=df2[,c("IID","norm_vitD")]
df2$FID=df2$IID
df2=df2[,c("FID","IID","norm_vitD")]
if(saveResults){write.table(df2, "input/mlm_pheno", row.names=F, quote=F)}Meta-analysis
To meta-analyse UKB results with results obtained by the SUNLIGHT consortium, we analysed the data in the same way, i.e.:
- Restricting data to individuals of European ancestry
- Using a log-transformed phenotype
- Including the following covariates in the model (fastGWA):
- Assessment month
- Age
- Sex
- BMI
- 40 PCs
- Genotyping batch
# Code from JennySeason-stratified GWAS
# Restrict to ALL individuals of European ancestry
df=read.table("input/vitaminD_tidyData_noMissing", h=T, stringsAsFactors=F)
df2=df
df2$FID=df2$IID
# Define seasons
## Winter = Dec - Apr
## Summer = Jun - Oct
df2$season=NULL
df2[df2$assessMonth<=4 | df2$assessMonth>=12,"season"]="winter"
df2[df2$assessMonth>=6 & df2$assessMonth<=10,"season"]="summer"
df2=df2[!is.na(df2$season),]
# stratify data and apply RINT
# Summarise
winter=df2[df2$season=="winter",c("FID","IID","vitaminD","norm_vitD","log_vitD")]
summer=df2[df2$season=="summer",c("FID","IID","vitaminD","norm_vitD","log_vitD")]
summary(summer$vitaminD); sd(summer$vitaminD); summary(winter$vitaminD); sd(winter$vitaminD)
#summary(summer$norm_vitD); sd(summer$norm_vitD); summary(winter$norm_vitD); sd(winter$norm_vitD)
#summary(summer$log_vitD); sd(summer$log_vitD); summary(winter$log_vitD); sd(winter$log_vitD)
winter=df2[df2$season=="winter",c("FID","IID","norm_vitD")]
summer=df2[df2$season=="summer",c("FID","IID","norm_vitD")]
norm = function(x){x[order(x,na.last=NA)]=qnorm(ppoints(length(x[!is.na(x)])),lower=T);x }
winter$norm_vitD=norm(winter$norm_vitD)
summer$norm_vitD=norm(summer$norm_vitD)
# Save "season" definition
df2$season_binary=ifelse(df2$season=="winter", 1, 2)
df2=df2[,c("FID","IID","norm_vitD","season","season_binary")]
if(saveResults){write.table(df2, "input/covariates_season", row.names=F, quote=F)}
# Save datasets to feed as phenotype to fastGWA
if(saveResults){write.table(winter, "input/mlm_winter_pheno", row.names=F, quote=F)}
if(saveResults){write.table(summer, "input/mlm_summer_pheno", row.names=F, quote=F)} After a visual inspection of the average vitamin D levels per month
(see Time series section), we defined two seasons of blood
draw and stratified the UKB sample into two cohorts based on those:
- “Winter” - individuals assessed Dec-Apr (N=162591)
- “Summer” - individuals assessed Jun-Oct (N=177082)
Individuals with vitamin D levels assessed in the months of May and November were not included in these analyses as the transition between seasons varies between years and could confound the results.
The two phenotype files (one for each season) were generated by (1) restricting to individuals of European ancestry, (2) restricting the individuals assessed in the defined season and (3) applying a rank-based inverse-normal transformation.
vQTL GWAS
The phenotype used in the variance quantitative trait loci (vQTL) GWAS was pre-regressed of confounder effects. Briefly, we:
- Restricted to unrelated European individuals
- Regressed-out the effect of confounders within the following groups:
- Females-vitD: females who ever reported taking vitamin D supplements (vitD group)
- Females-other: females who ever reported taking supplements, but never vitamin D (other group)
- Females-none: females who always reported not taking supplements (none group)
- Females-NA females with no information on supplement intake (NA group)
- Males-vitD: males who ever reported taking vitamin D supplements (vitD group)
- Males-other: males who ever reported taking supplements, but never vitamin D other group)
- Males-none: males who always reported not taking supplements (none group)
- Males-NA: males with no information on supplement intake NA group)
- Removed outliers more than 5 SD from the mean
- Standardised the residuals to get mean 0 and SD 1 (note that this is not a non-linear transformation and will help with interpretation of downstream analyses)
- Merged residuals from the different groups
Variables regressed from the phenotype were:
- Age
- Assessment month
- Assessment centre
- First 40 PCs
- Genotyping batch
# Add PCs to our data frame to remove their effect
df=read.table("input/vitaminD_tidyData_noMissing", h=T, stringsAsFactors=F)
#load("input/ukbEUR_PC40_HM3excludeRegions_v2.RData")
df2=merge(df, pcs, all.x=T)
# Restrict to unrelated individuals of European ancestry
v3EURu_HM3_IDs=read.table("input/ukbEURu_imp_chr1_v3_HM3.fam", h=F, stringsAsFactors=F)
#nrow(v3EURu_HM3_IDs)
names(v3EURu_HM3_IDs)[1:2]=c("FID","IID")
df2=df2[df2$IID %in% v3EURu_HM3_IDs$IID,]
#length(df2$IID)There were 348,501 unrelated individuals of European ancestry. Of those, 318,851 had vitamin D levels assessed and will be analysed in this preliminary analysis.
# Show sample size of the groups within which we will run covariate regression
tmp=table(df2$sex, df2$supplementsEver, exclude=NULL)
tmp=kable(tmp, caption="Sample size of groups within which we ran covariate regression")
t1=kable_styling(tmp, full_width=F)
# Define the groups within which we will run covariate regression
df2[is.na(df2$supplementsEver),"supplementsEver"]="NA"
df2$group=paste(df2$sex, df2$supplementsEver, sep="_")
# Code categorical variables as factors
df2[,c("assessCentre","assessMonth","batch")] = lapply(df2[,c("assessCentre","assessMonth","batch")], factor)
# Regress effect of covariates
nPCs=40 # number of PCs to include
for(i in unique(df2$group))
{
# Get residuals
test=paste("vitaminD ~ age + assessCentre + assessMonth + batch +", paste(paste0("PC",1:nPCs),collapse = "+"))
model=lm(as.formula(test), df2[df2$group==i,])
res=resid(model)
# Remove outliers (>5 SD from mean)
low=mean(res)-5*sd(res)
high=mean(res)+5*sd(res)
res[res<low | res>high]=NA
# Standardize residuals for mean 0 and SD 1 (note: this is not a non-lienar transformation and will help with the interpretation of the results)
res_std=(res-mean(res, na.rm=T))/sd(res, na.rm=T)
# Add standardized residuals to data frame
df2[df2$group==i,"vitD_res"]=res_std
}
df2=df2[!is.na(df2$vitD_res),]
# Plot residuals by group
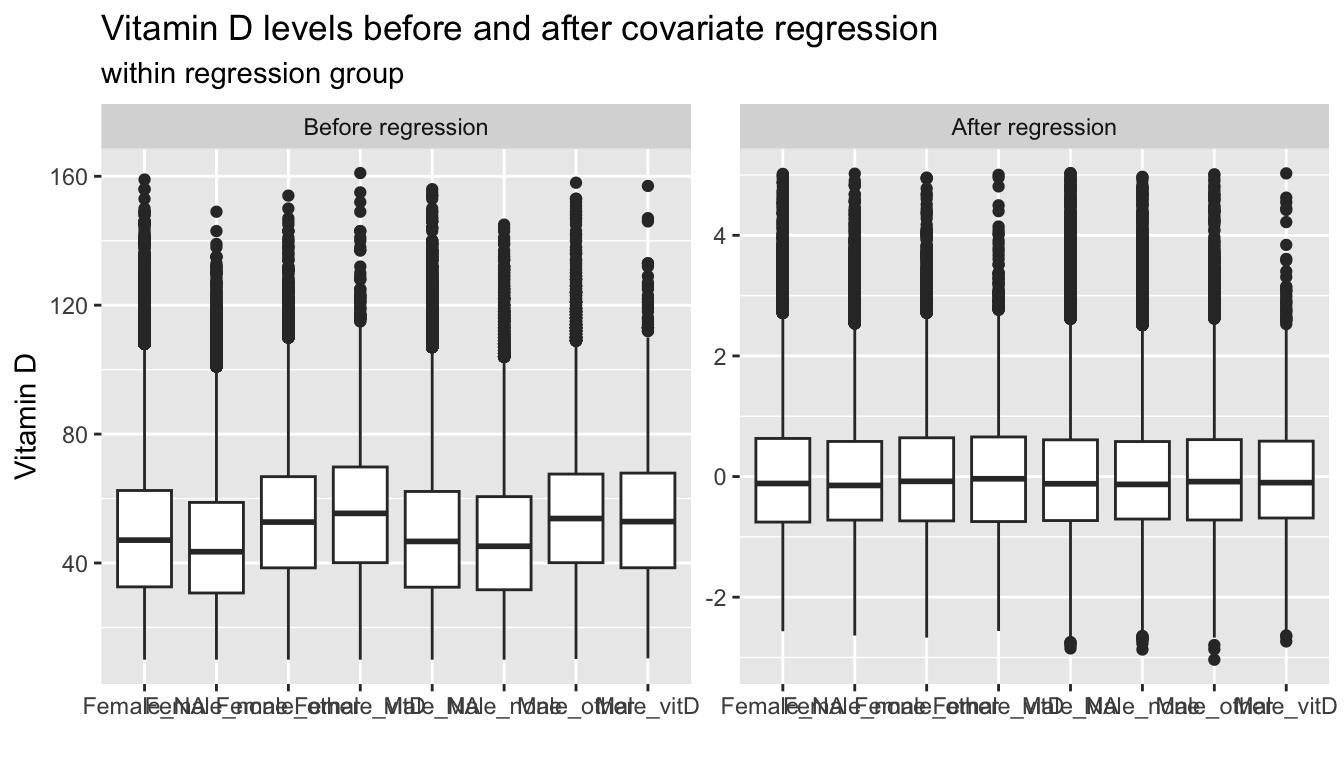
tmp=reshape2::melt(df2[,c("IID","vitaminD","vitD_res","group")], id.vars=c("IID", "group"))
tmp$variable=factor(tmp$variable, levels=c("vitaminD","vitD_res"), labels=c("Before regression","After regression"))
p1=ggplot(tmp, aes(x=group, y=value)) +
geom_boxplot() +
facet_wrap(variable~., scales="free") +
labs(title="Vitamin D levels before and after covariate regression",
subtitle="within regression group",
x="",y="Vitamin D")
# Plot adjusted phenotype
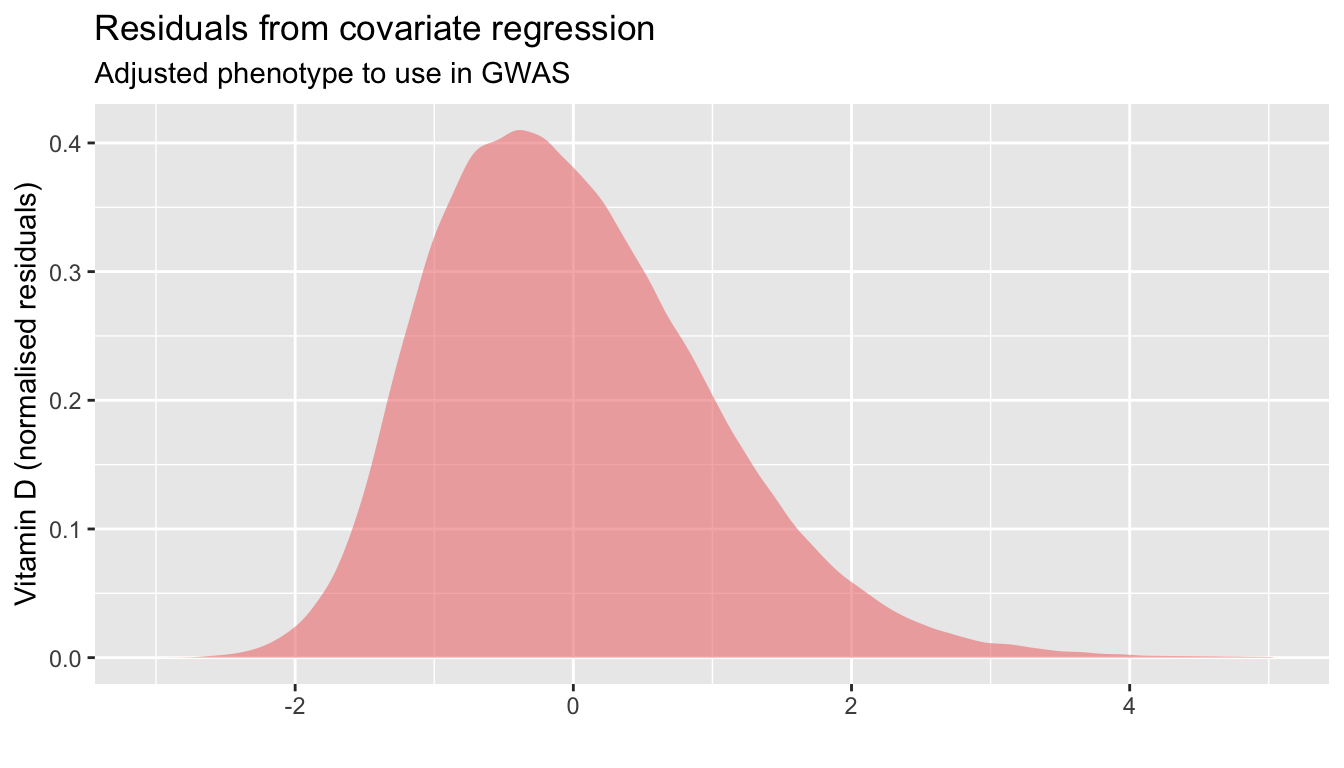
p2=ggplot(df2, aes(x=vitD_res)) +
geom_density(fill="lightcoral", color=alpha("grey", 0),alpha = 0.6) +
labs(title="Residuals from covariate regression",
subtitle="Adjusted phenotype to use in GWAS",
x="",y="Vitamin D (normalised residuals)")
# Save residuals to feed as phenotype for the vQTL GWAS
df2=df2[,c("IID","vitD_res")]
df2$FID=df2$IID
df2=df2[,c("FID","IID","vitD_res")]
if(saveResults){write.table(df2, "input/vqtl_pheno", row.names=F, quote=F)}
# Save env
vars2keep=c("p1","p2","t1")
save(list=vars2keep, file = "rdm/input/rmd/vqtl_cov_regression.RData")
t1;p1;p2| none | other | vitD | NA | |
|---|---|---|---|---|
| Female | 34278 | 35309 | 6624 | 93901 |
| Male | 36969 | 24897 | 2655 | 84218 |