Vitamin D - Heritability
To estimate the proportion of phenotypic variability explained by our meta-analysed GWAS results, we conducted the following analyses:
- LD score regression
- GRP: we performed an out-of-sample genomic risk prediction (GRP) analysis into an independent dataset (QIMR cohort; N=6,223)
- GREML: we used the genomic-relatedness-based restricted maximum-likelihood (GREML) approach implemented in GCTA to estimate the variability explained in a sub-sample of unrelated 50K individuals from the UKB
In addition, we estimated the proportion of variance explained by the variants with strongest association with vitamin D levels by conducting a GREML analysis in the QIMR cohort using the list of independent variants identified with COJO.
LDSC
module load ldsc
prefix=vitD_fastGWA_sunlight_meta.ukbSNPs
# Directories
ldscDir=$bin/ldsc
results=$WD/results/sunlight
tmpDir=$results/tmpDir
# Format GWAS results
awk '{print $1,$2,$3,$5,$8,$7}' $results/$prefix.ma > $tmpDir/$prefix.ldHubFormat
# Restrict to common SNPs
sed 1q $tmpDir/$prefix.ldHubFormat > $results/$prefix.ldHubFormat
awk 'NR==FNR{a[$1];next} ($1 in a)' $WD/input/UKB_v3EURu_impQC/UKB_EURu_impQC_maf0.01.snplist <(awk 'NR==FNR{a[$1];next} ($1 in a)' $WD/input/w_hm3.noMHC.snplist $tmpDir/$prefix.ldHubFormat) >> $results/$prefix.ldHubFormat
# Munge sumstats (process sumstats and restrict to HapMap3 SNPs)
munge_sumstats.py --sumstats $results/$prefix.ldHubFormat \
--merge-alleles $ldscDir/w_hm3.snplist \
--out $tmpDir/$prefix
# Run LDSC
ldsc.py --h2 $tmpDir/$prefix.sumstats.gz \
--ref-ld-chr $ldscDir/eur_w_ld_chr/ \
--w-ld-chr $ldscDir/eur_w_ld_chr/ \
--out $results/$prefix.ldsc
# Copy results to local computer (run command in local computer)
scp $deltaID:$WD/results/sunlight/vitD_fastGWA_sunlight_meta.ukbSNPs.ldsc.log $local/results/sunlight/tmp=read.table("results/fastGWA/ldsc/vitD_fastGWA.ldsc.log", skip=15, fill=T, h=F, colClasses="character", sep="_")[,1]
# Get results:
h2=strsplit(grep("Total Observed scale h2",tmp, value=T),":")[[1]][2]
lambda_gc=strsplit(grep("Lambda GC",tmp, value=T),":")[[1]][2]
chi2=strsplit(grep("Mean Chi",tmp, value=T),":")[[1]][2]
intercept=strsplit(grep("Intercept",tmp, value=T),":")[[1]][2]
ratio=strsplit(grep("Ratio",tmp, value=T),":")[[1]][2]We ran LDSC on our GWAS results (obtained with fastGWA). Results were:
- Total Observed scale h2: 0.0966 (0.0138)
- Lambda GC: 1.4817
- Mean Chi2: 1.881
- Intercept: 1.0628 (0.0136)
- Ratio: 0.0713 (0.0154)
GRP
GRP was conducted by the QIMR group using our GWAS results (N=417580; M=8806780). The following covariates were regressed from the phenotype (25OHD):
- sex
- age
- month of collection
- 10PCs
- imputation run
Then, residuals were log-transformed.
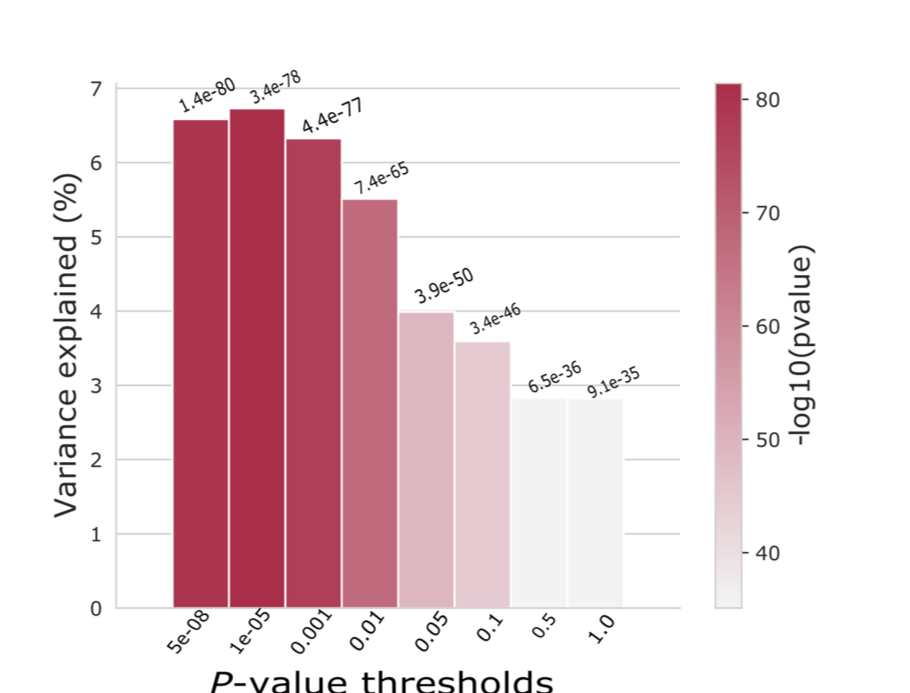
The plot below summarises the results of GRP conducted with different p-value thresholds:
module load ldsc
prefix=vitD_qimrGWAS
# Directories
ldscDir=$bin/ldsc
results=$WD/results/h2
tmpDir=$results/tmpDir
# Restrict to common SNPs
echo 'args=commandArgs(trailingOnly=T)
gwasSumstats=args[1]
out=args[2]
df=read.table(gwasSumstats,h=F,stringsAsFactors=F)
df=df[df[,4]>1e-6 & df[,10]>.01,]
write.table(df, out, quote=F, row.names=F)' > $WD/scripts/restrictQIMR_hwe1e6.maf0.01
cd $results
tmp_command="Rscript $WD/scripts/restrictQIMR_hwe1e6.maf0.01 $WD/input/qimr/hrc.rw1.use.gz $tmpDir/$prefix.hwe1e6.maf0.01"
qsubshcom "$tmp_command" 1 10G restrictQIMR 5:00:00 ""
# Format GWAS results
echo "SNP A1 A2 b n p" > $results/$prefix.ldHubFormat
awk '{print $2,$8,$9,$11,$14,$13}' $tmpDir/$prefix.hwe1e6.maf0.01 | sed 1d>> $results/$prefix.ldHubFormat
# Munge sumstats (process sumstats and restrict to HapMap3 SNPs)
munge_sumstats.py --sumstats $results/$prefix.ldHubFormat \
--merge-alleles $ldscDir/w_hm3.snplist \
--out $tmpDir/$prefix
# Run LDSC
ldsc.py --h2 $tmpDir/$prefix.sumstats.gz \
--ref-ld-chr $ldscDir/eur_w_ld_chr/ \
--w-ld-chr $ldscDir/eur_w_ld_chr/ \
--out $results/$prefix.ldsctmp=read.table("results/h2/vitD_qimrGWAS.ldsc.log", skip=15, fill=T, h=F, colClasses="character", sep="_")[,1]
# Get results:
h2=strsplit(grep("Total Observed scale h2",tmp, value=T),":")[[1]][2]
lambda_gc=strsplit(grep("Lambda GC",tmp, value=T),":")[[1]][2]
chi2=strsplit(grep("Mean Chi",tmp, value=T),":")[[1]][2]
intercept=strsplit(grep("Intercept",tmp, value=T),":")[[1]][2]
ratio=strsplit(grep("Ratio",tmp, value=T),":")[[1]][2]We ran LDSC on the QIMR GWAS results to estimate the proportion of phenotypic variance explained with those results:
- Total Observed scale h2: -0.2195 (0.0819)
- Lambda GC: 1.0016
- Mean Chi2: 1.0109
- Intercept: 1.038 (0.006)
- Ratio: 3.4737 (0.5455)
The negative h2 estimate was accompanied by a low mean Chi2 (1.011), indicating that results from this GWAS are not polygenic enough to run LDSC (refer to LDSC FAQs). This could be explained by the small sample size of the QIMR cohort (N=6,223, including related individuals)
GREML
Out-of-sample GREML
To get an unbiased estimate the phenotypic variability explained by the independent SNPs in our GWAS results, we conducted a GREML analysis on the QIMR dataset using independent variants identified with COJO. First, we restricted the UKB GWAS results to the variants that were also assessed in the QIMR cohort. Second, we ran COJO on the restricted results. Third, we conducted a GREML analysis on the QIMR data using the set of independent variants identified with COJO.
COJO
# Load GWAS results
if(F)
{
# Read results
qimr=read.table("input/qimr/hrc.rw1.use.gz", h=F, stringsAsFactors=F)
# Add variable names
tmp=read.table("input/qimr/hrc.rw1.use.head.txt", h=T, fill=T)
names(qimr)=names(tmp)
# Save as RDS
saveRDS(qimr, "input/qimr/vitD_qimrGWAS.RDS")
}
qimr=readRDS("input/qimr/vitD_qimrGWAS.RDS")
# Generate list of SNPs to use in COJO (i.e. variants in QIMR dataset and in the list of variants that we usually include in COJO analyses)
cojo_snps=read.table("input/UKB_v3EUR_impQC/UKB_EUR_impQC_maf0.01.snplist", h=F, stringsAsFactors=F, col.names="SNP")
tmp=cojo_snps[cojo_snps$SNP %in% qimr$DBSNP,]
write.table(tmp, "input/UKB_v3EUR_impQC/UKB_EUR_impQC_maf0.01.qimrSNPs.snplist", quote=F, row.names=F, col.names=F)QIMR GWAS results were available for 7676006 variants. We restricted the UKB GWAS results to these variants and ran COJO with the same settings as before:
- –bfile: For autosomes, we used the GWAS individual-level genotype data (
v3EUR_impQC) as LD reference. For the X chromosome, we used v3EUR_impQC female samples. - –cojo-slct: Performs a step-wise model selection of independently associated SNPs
- –cojo-p 5e-8 (default): P-value to declare a genome-wide significant hit
- –cojo-wind 10,000 (default): Distance (in KB) from which SNPs are considered in LD
- –cojo-collinear 0.9 (default): maximum r2 to be considered an independent SNP, i.e. SNPs with r2>0.9 will not be considered further
- –diff-freq 0.2 (default): maximum allele difference bet
- extract: Restrict analysis to SNPs with MAF>0.01
# #########################################
# Run COJO on GWAS results - autosomes
# #########################################
# Directories
results=$WD/results/h2
tmpDir=$results/tmpDir
# Files/settings to use
prefix=vitD_fastGWA.qimrSNPs
random20k_dir=$WD/input/UKB_v3EURu_impQC/random20K
ldRef=$random20k_dir/ukbEURu_imp_chr{TASK_ID}_v3_impQC_random20k_maf0.01
filters=maf0.01
inFile=$WD/results/fastGWA/maFormat/vitD_fastGWA.ma
snps2include=$WD/input/UKB_v3EUR_impQC/UKB_EUR_impQC_$filters.qimrSNPs.snplist
outFile=$tmpDir/${prefix}_${filters}_chr{TASK_ID}
# Run COJO
cd $results
gcta=$bin/gcta/gcta_1.92.3beta3
tmp_command="$gcta --bfile $ldRef \
--cojo-slct \
--cojo-file $inFile \
--extract $snps2include \
--out ${outFile}"
qsubshcom "$tmp_command" 1 100G $prefix.COJO 30:00:00 "-array=1-22"
# #########################################
# Run COJO on GWAS results - X chromosome
# #########################################
# Directories
results=$WD/results/h2
tmpDir=$results/tmpDir
# Generate list of QIMR SNPs
#zcat $WD/input/qimr/hrc.rw1.use.gz | awk '{print $2}' > $WD/input/qimr/qimr.snplist
# Files/settings to use
prefix=vitD_fastGWA.qimrSNPs
snps2include=$WD/input/qimr/qimr.snplist
maf=0.01
filters=maf$maf
# Run COJO
cd $results
gcta=$bin/gcta/gcta_1.92.3beta3
for chr in X XY
do
ref=$medici/v3EUR_chrX/ukbEURv3_${chr}_maf0.1_FEMALE
inFile=$WD/results/fastGWA/maFormat/vitD_fastGWA_chr$chr.ma
outFile=$tmpDir/${prefix}_maf${maf}_chr$chr
tmp_command="$gcta --bfile $ref \
--cojo-slct \
--cojo-file $inFile \
--maf $maf \
--extract $snps2include \
--out ${outFile}"
qsubshcom "$tmp_command" 1 30G $prefix.chr$chr.COJO 24:00:00 ""
done
# #########################################
# Merge COJO results once all chr finished running
# #########################################
cat $tmpDir/${prefix}_${filters}*.jma.cojo | grep "Chr" | uniq > $results/${prefix}_${filters}_withX.cojo
cat $tmpDir/${prefix}_${filters}_chr*.jma.cojo | grep -v "Chr" |sed '/^$/d' >> $results/${prefix}_${filters}_withX.cojo
cojo=read.table("results/h2/vitD_fastGWA.qimrSNPs_maf0.01_withX.cojo", h=T, stringsAsFactors=F)We identified 143 independent variants to use run GREML on with the QIMR individual-level data.
Clump
The out-of-sample prediction with COJO variants (r2=0.1052, P=2.85x10-134) was better than that for GWS (P<5x10-8) SNPs (r2=0.072, P=5.95x10-88). To check that this is right, we restricted the UKB results to SNPs in the QIMR data set, clumped them and performed out-of-sample prediction with GWS variants.
# #########################################
# Clump GWAS results - autossomes
# #########################################
# Directories
results=$WD/results/h2
tmpDir=$results/tmpDir
# Files/settings to use
prefix=vitD_fastGWA.qimrSNPs
random20k_dir=$WD/input/UKB_v3EURu_impQC/random20K
ldRef=$random20k_dir/ukbEURu_imp_chr{TASK_ID}_v3_impQC_random20k_maf0.01
snps2exclude=$WD/input/UKB_v3EUR_impQC/UKB_EUR_impQC_duplicated_rsIDs
filters=maf0.01
snps2include=$WD/input/UKB_v3EUR_impQC/UKB_EUR_impQC_$filters.qimrSNPs.snplist
inFile=$WD/results/fastGWA/maFormat/vitD_fastGWA.ma
window=10000
outFile=$tmpDir/${prefix}_chr{TASK_ID}
jobName=$prefix.clump
# Run clumping
cd $results
plink=$bin/plink/plink-1.9/plink
tmp_command="$plink --bfile $ldRef \
--exclude $snps2exclude \
--extract $snps2include \
--clump $inFile \
--clump-p1 5e-8 \
--clump-r2 0.01 \
--clump-kb $window \
--clump-field "p" \
--out $outFile"
qsubshcom "$tmp_command" 1 30G $jobName 24:00:00 "-array=1-22"
# #########################################
# Clump GWAS results - chromosome X
# #########################################
# Directories
results=$WD/results/h2
tmpDir=$results/tmpDir
# Run clumping
cd $results
for chr in X XY
do
# Files/settings to use
prefix=vitD_fastGWA.qimrSNPs_chr$chr
ldRef=$medici/v3EUR_chrX/ukbEURv3_${chr}_maf0.1_FEMALE
inFile=$WD/results/fastGWA/maFormat/vitD_fastGWA_chr$chr.ma
snps2include=$WD/input/qimr/qimr.snplist
outFile=$tmpDir/$prefix
plink=$bin/plink/plink-1.9/plink
tmp_command="$plink --bfile $ldRef \
--extract $snps2include \
--clump $inFile \
--clump-p1 5e-8 \
--clump-r2 0.01 \
--clump-kb 10000 \
--clump-field "p" \
--out $outFile"
qsubshcom "$tmp_command" 1 10G $prefix.clump 10:00:00 ""
echo $prefix.clump
done
# #########################################
# Merge clumped results once all chr finished running
# #########################################
cat $tmpDir/${prefix}_chr*clumped | grep "CHR" | head -1 > $results/${prefix}_withX.clumped
cat $tmpDir/${prefix}_chr*clumped | grep -v "CHR" |sed '/^$/d' >> $results/${prefix}_withX.clumped
# ############################
# Restrict UKB sumstats to this list of SNPs to built PRS for prediction into QIMR
# ############################
# Ran locally in R
gwas=readRDS("results/fastGWA/vitD_fastGWA_withX.RDS")
clump=read.table("results/h2/vitD_fastGWA.qimrSNPs_withX.clumped", h=T, stringsAsFactors=T)
tmp=gwas[gwas$SNP %in% clump$SNP,]
write.table(tmp, "results/fastGWA/tmpDir/vitD_fastGWA_withX_restricted2QIMR.clumped", row.names=F, quote=F)GREML results
GREML was conducted with the 143 independent variants identified with COJO. 75 SNPs had been directly genotyped in the QIMR data set. All other SNPs were imputed with imputation quality > 0.8. GREML was conducted using a subset of 1,632 unrelated individuals (taking one person from each family).
- V(G)/V(P) of 0.1 (SE 0.02)
Note: These results are still based on the 213 COJO variants that we sent in Aug - needs updating